Learning structure-energy relations
Machine learning in computational chemistry has emerged as an indispensable driving force for innovation. By combining the low cost of traditional empirical potentials with the accuracy of ab initio methods, machine learning potentials deliver long time and large length scales required for insight into complex molecular systems. Instead of physically motivated forms of interactions, the structure-energy relation is represented by highly flexible mathematical functions, such as neural networks or kernel based approaches, which are trained to reproduce high level reference calculations. Main challenges in the field are the generation of robust and accurate models, but also the construction of representative training data. Our group uses neural network based approaches and has a strong background in automating the generation of training data to provide robust models.
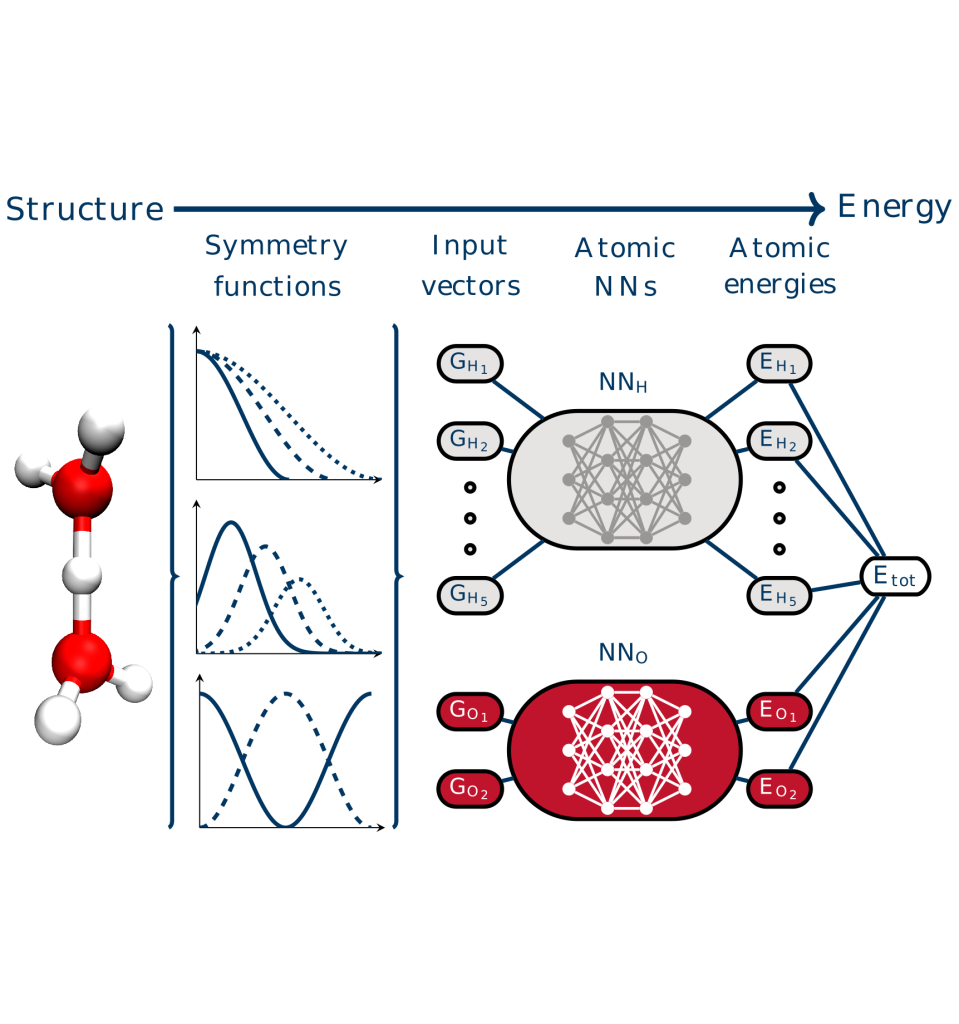
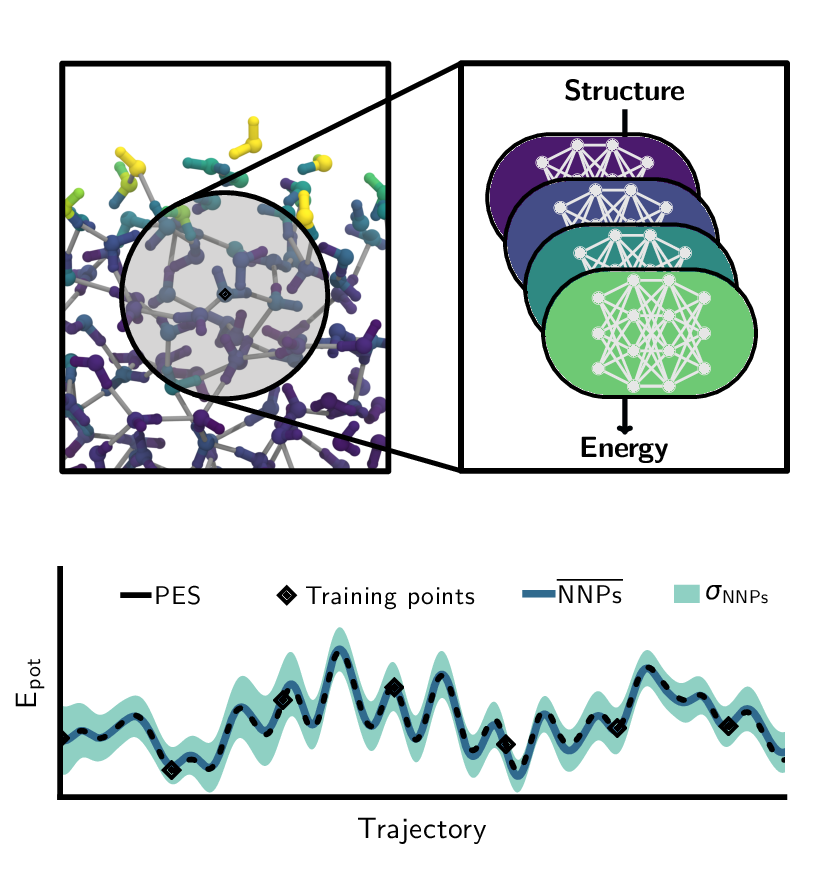
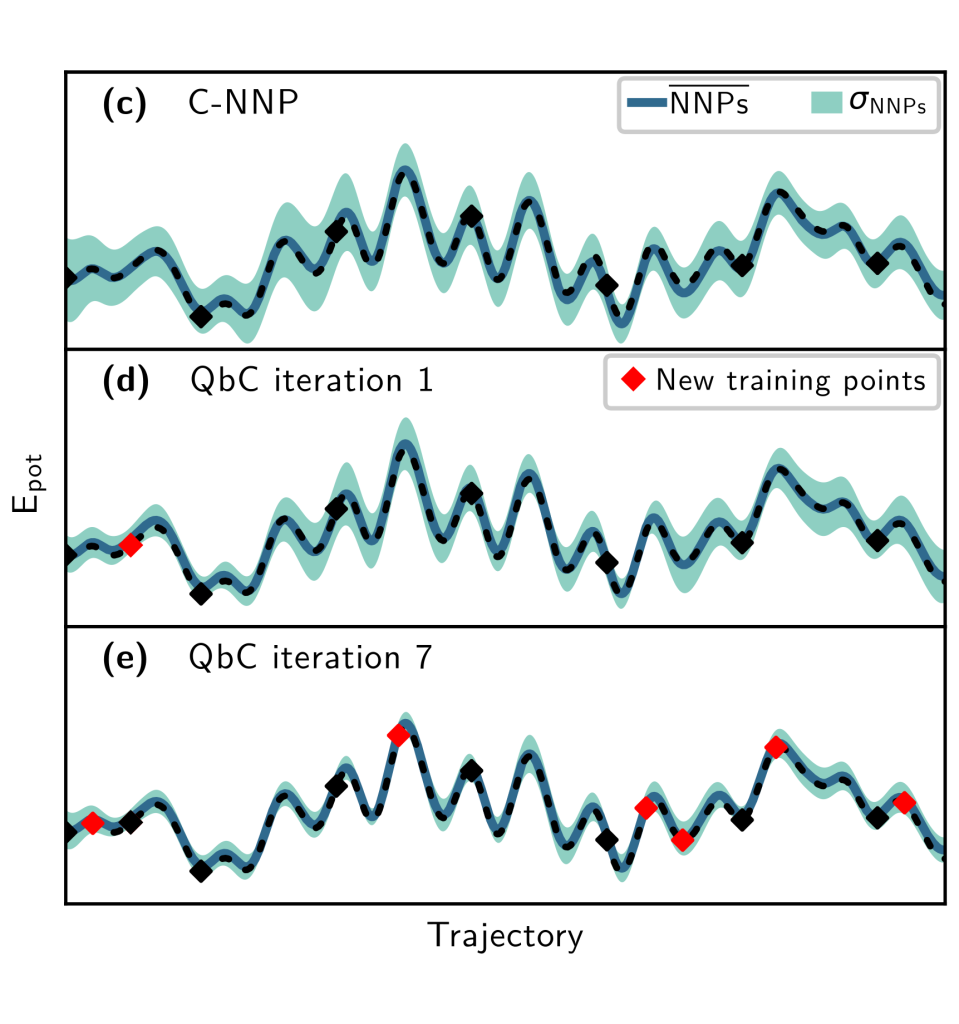
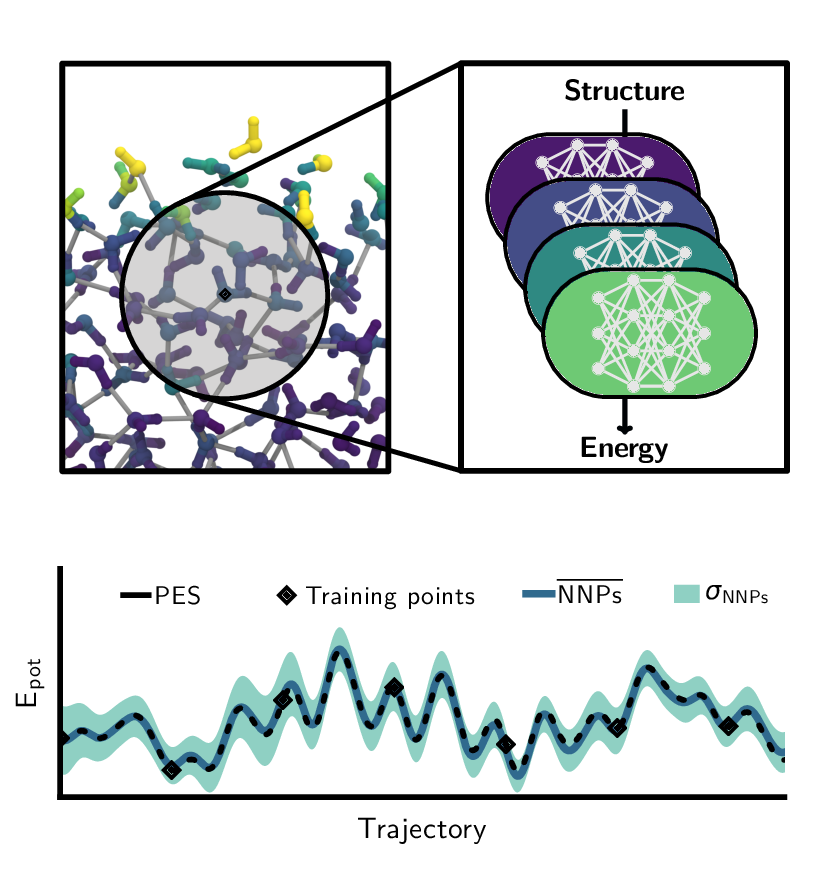
Overview of the machine learning approaches used in our research. Left: Architecture of neural network potentials. Middle: Schematics of committee neural network potentials. Right: Concept of the active learning technique query by committee.
The recent advances in the field of machine learning potentials have only now opened up the door to investigate many challenging phenomena with great prospect for future work.[1][2][3]
References
- (2020): Automated Fitting of Neural Network Potentials at Coupled Cluster Accuracy: Protonated Water Clusters as Testing Ground. In: J. Chem. Theory Comput., vol. 16, no. 1, pp. 88–99, 2020, ISSN: 15499626.
- (2020): Committee neural network potentials control generalization errors and enable active learning. In: J. Chem. Phys., vol. 153, no. 10, pp. 104105, 2020, ISSN: 10897690.
- (2021): Machine learning potentials for complex aqueous systems made simple. In: Proc. Natl. Acad. Sci., vol. 118, no. 38, pp. e2110077118, 2021, ISSN: 0027-8424.