2025

Ilyes Batatia, Philipp Benner, Yuan Chiang, Alin M. Elena, Dávid P. Kovács, Janosh Riebesell, Xavier R. Advincula, Mark Asta, Matthew Avaylon, William J. Baldwin, Fabian Berger, Noam Bernstein, Arghya Bhowmik, Filippo Bigi, Samuel M. Blau, Vlad Cărare, Michele Ceriotti, Sanggyu Chong, James P. Darby, Sandip De, Flaviano Della Pia, Volker L. Deringer, Rokas Elijošius, Zakariya El-Machachi, Edvin Fako, Fabio Falcioni, Andrea C. Ferrari, John L.A. Gardner, Mikołaj J. Gawkowski, Annalena Genreith-Schriever, Janine George, Rhys E.A. Goodall, Jonas Grandel, Clare P. Grey, Petr Grigorev, Shuang Han, Will Handley, Hendrik H. Heenen, Kersti Hermansson, Cheuk Hin Ho, Stephan Hofmann, Christian Holm, Jad Jaafar, Konstantin S. Jakob, Hyunwook Jung, Venkat Kapil, Aaron D. Kaplan, Nima Karimitari, James R. Kermode, Panagiotis Kourtis, Namu Kroupa, Jolla Kullgren, Matthew C. Kuner, Domantas Kuryla, Guoda Liepuoniute, Chen Lin, Johannes T. Margraf, Ioan-Bogdan Magdău, Angelos Michaelides, J. Harry Moore, Aakash A. Naik, Samuel P. Niblett, Sam Walton Norwood, Niamh O’Neill, Christoph Ortner, Kristin A. Persson, Karsten Reuter, Andrew S. Rosen, Louise A.M. Rosset, Lars L. Schaaf, Christoph Schran, Benjamin X. Shi, Eric Sivonxay, Tamás K. Stenczel, Christopher Sutton, Viktor Svahn, Thomas D. Swinburne, Jules Tilly, Cas van der Oord, Santiago Vargas, Eszter Varga-Umbrich, Tejs Vegge, Martin Vondrák, Yangshuai Wang, William C. Witt, Thomas Wolf, Fabian Zills, Gábor Csányi
A foundation model for atomistic materials chemistry Journal Article
In: J. Chem. Phys., vol. 163, iss. 18, pp. 184110, 2025.
Abstract | Links | BibTeX | Tags: Machine Learning Potentials
@article{Batatia2025/10.1063/5.0297006,
title = {A foundation model for atomistic materials chemistry},
author = {Ilyes Batatia and Philipp Benner and Yuan Chiang and Alin M. Elena and Dávid P. Kovács and Janosh Riebesell and Xavier R. Advincula and Mark Asta and Matthew Avaylon and William J. Baldwin and Fabian Berger and Noam Bernstein and Arghya Bhowmik and Filippo Bigi and Samuel M. Blau and Vlad Cărare and Michele Ceriotti and Sanggyu Chong and James P. Darby and Sandip De and Flaviano Della Pia and Volker L. Deringer and Rokas Elijošius and Zakariya El-Machachi and Edvin Fako and Fabio Falcioni and Andrea C. Ferrari and John L.A. Gardner and Mikołaj J. Gawkowski and Annalena Genreith-Schriever and Janine George and Rhys E.A. Goodall and Jonas Grandel and Clare P. Grey and Petr Grigorev and Shuang Han and Will Handley and Hendrik H. Heenen and Kersti Hermansson and Cheuk Hin Ho and Stephan Hofmann and Christian Holm and Jad Jaafar and Konstantin S. Jakob and Hyunwook Jung and Venkat Kapil and Aaron D. Kaplan and Nima Karimitari and James R. Kermode and Panagiotis Kourtis and Namu Kroupa and Jolla Kullgren and Matthew C. Kuner and Domantas Kuryla and Guoda Liepuoniute and Chen Lin and Johannes T. Margraf and Ioan-Bogdan Magdău and Angelos Michaelides and J. Harry Moore and Aakash A. Naik and Samuel P. Niblett and Sam Walton Norwood and Niamh O’Neill and Christoph Ortner and Kristin A. Persson and Karsten Reuter and Andrew S. Rosen and Louise A.M. Rosset and Lars L. Schaaf and Christoph Schran and Benjamin X. Shi and Eric Sivonxay and Tamás K. Stenczel and Christopher Sutton and Viktor Svahn and Thomas D. Swinburne and Jules Tilly and Cas van der Oord and Santiago Vargas and Eszter Varga-Umbrich and Tejs Vegge and Martin Vondrák and Yangshuai Wang and William C. Witt and Thomas Wolf and Fabian Zills and Gábor Csányi},
url = {https://pubs.aip.org/aip/jcp/article/163/18/184110/3372267},
doi = {10.1063/5.0297006},
year = {2025},
date = {2025-11-13},
journal = {J. Chem. Phys.},
volume = {163},
issue = {18},
pages = {184110},
abstract = {Atomistic simulations of matter, especially those that leverage first-principles (ab initio) electronic structure theory, provide a microscopic view of the world, underpinning much of our understanding of chemistry and materials science. Over the last decade or so, machine-learned force fields have transformed atomistic modeling by enabling simulations of ab initio quality over unprecedented time and length scales. However, early machine-learning (ML) force fields have largely been limited by (i) the substantial computational and human effort required to develop and validate potentials for each particular system of interest and (ii) a general lack of transferability from one chemical system to the next. Here, we show that it is possible to create a general-purpose atomistic ML model, trained on a public dataset of moderate size, that is capable of running stable molecular dynamics for a wide range of molecules and materials. We demonstrate the power of the MACE-MP-0 model—and its qualitative and at times quantitative accuracy—on a diverse set of problems in the physical sciences, including properties of solids, liquids, gases, chemical reactions, interfaces, and even the dynamics of a small protein. The model can be applied out of the box as a starting or “foundation” model for any atomistic system of interest and, when desired, can be fine-tuned on just a handful of application-specific data points to reach ab initio accuracy. Establishing that a stable force-field model can cover almost all materials changes atomistic modeling in a fundamental way: experienced users obtain reliable results much faster, and beginners face a lower barrier to entry. Foundation models thus represent a step toward democratizing the revolution in atomic-scale modeling that has been brought about by ML force fields.},
keywords = {Machine Learning Potentials},
pubstate = {published},
tppubtype = {article}
}

Niamh O’Neill, Benjamin X. Shi, William J. Baldwin, William C. Witt, Gábor Csányi, Julian D. Gale, Angelos Michaelides, Christoph Schran
Towards routine condensed phase simulations with delta-learned coupled cluster accuracy: Application to liquid water Journal Article
In: J. Chem. Theory Comput., 2025.
Abstract | Links | BibTeX | Tags: Coupled Cluster, Machine Learning Potentials, Water
@article{ONeill2025/10.1021/acs.jctc.5c01377,
title = {Towards routine condensed phase simulations with delta-learned coupled cluster accuracy: Application to liquid water},
author = {Niamh O’Neill and Benjamin X. Shi and William J. Baldwin and William C. Witt and Gábor Csányi and Julian D. Gale and Angelos Michaelides and Christoph Schran},
url = {https://pubs.acs.org/doi/full/10.1021/acs.jctc.5c01377},
doi = {10.1021/acs.jctc.5c01377},
year = {2025},
date = {2025-11-07},
journal = {J. Chem. Theory Comput.},
abstract = {Simulating liquid water to an accuracy that matches its wealth of available experimental data requires both precise electronic structure methods and reliable sampling of nuclear (quantum) motion. This is challenging because applying the electronic structure method of choice, coupled cluster theory with single, double, and perturbative triple excitations [CCSD(T)] to condensed phase systems, is currently limited by its computational cost and complexity. Recent tour de force efforts have demonstrated that this accuracy can indeed bring simulated liquid water into close agreement with experiment using machine learning potentials (MLPs). However, achieving this remains far from routine, requiring large datasets and significant computational cost. In this work, we introduce a practical approach that combines developments in MLPs with local correlation approximations to enable routine CCSD(T)-level simulations of liquid water. When combined with nuclear quantum effects, we achieve agreement with experiments for structural and transport properties. Importantly, the approach also handles constant-pressure simulations, enabling MLP-based CCSD(T) models to predict isothermal–isobaric bulk properties, such as water’s density maximum, in close agreement with experiment. Encompassing tests across electronic structure, datasets, and MLP architecture, this work provides a practical blueprint towards routinely developing CCSD(T)-based MLPs for the condensed phase.},
keywords = {Coupled Cluster, Machine Learning Potentials, Water},
pubstate = {published},
tppubtype = {article}
}

Zeke Coady, Samuel G.H. Brookes, Zhaohan Shen, Benjamin J. Rhodes, Grace Mapstone, Zhen Xu, Wei Yu, Hirotomo Nishihara, Christoph Schran, Angelos Michaelides, Alexander C. Forse
Unexpected oversolubility of CO$_2$ measured at electrode–electrolyte interfaces Journal Article
In: J. Am. Chem. Soc., vol. 147, iss. 40, pp. 36310–36319, 2025.
Abstract | Links | BibTeX | Tags: Machine Learning Potentials, Water at Interfaces
@article{Coady2025/10.1021/jacs.5c09712,
title = {Unexpected oversolubility of CO$_2$ measured at electrode–electrolyte interfaces},
author = {Zeke Coady and Samuel G.H. Brookes and Zhaohan Shen and Benjamin J. Rhodes and Grace Mapstone and Zhen Xu and Wei Yu and Hirotomo Nishihara and Christoph Schran and Angelos Michaelides and Alexander C. Forse},
url = {https://pubs.acs.org/doi/full/10.1021/jacs.5c09712},
doi = {10.1021/jacs.5c09712},
year = {2025},
date = {2025-09-23},
urldate = {2025-09-23},
journal = {J. Am. Chem. Soc.},
volume = {147},
issue = {40},
pages = {36310–36319},
abstract = {Enhancements in gas solubility in pore-confined liquids─termed oversolubility─can drastically influence gas separation and catalytic efficiency in confined environments; however, they remain poorly understood in electrochemical $CO_2$ capture and reduction systems. While previous investigations of oversolubility have emphasized the importance of mesoporosity and incomplete pore saturation by the solvent, in this work, we report an unprecedented 30-fold oversolubility effect for $CO_2$ in solely microporous activated carbons saturated with 1 M $Na_2SO_{4(aq)}$. The oversolubility effect occurs regardless of the activated carbon’s functional groups and level of disorder and is enhanced for smaller pore sizes. Oversolubility is quantified using solid-state $^{13}C$ nuclear magnetic resonance spectroscopy (NMR), enabling differentiation between in-pore and ex-pore $CO_2$ and $HCO_3^–$. Atomistic modeling of the system, based on a machine-learning model delivering first-principles accuracy, suggests that the effect is driven by an adsorption-like mechanism underpinned by favorable interactions between $CO_2$ and the pore walls. Our findings demonstrate the unexpected importance of oversolubility for gas uptake in microporous, solvent-saturated carbon electrodes, an effect with direct relevance for improving electrochemical $CO_2$ capture and conversion technologies.},
keywords = {Machine Learning Potentials, Water at Interfaces},
pubstate = {published},
tppubtype = {article}
}

Samuel G.H. Brookes, Venkat Kapil, Angelos Michaelides, Christoph Schran
CO$_2$ hydration at the air–water interface: A surface-mediated “in-and-out” mechanism Journal Article
In: Proc. Natl. Acad. Sci., vol. 122, no. 34, pp. e2502684122, 2025.
Abstract | Links | BibTeX | Tags: Machine Learning Potentials, Water at Interfaces
@article{Brookes2025/10.1073/pnas.2502684122,
title = {CO$_2$ hydration at the air–water interface: A surface-mediated “in-and-out” mechanism},
author = {Samuel G.H. Brookes and Venkat Kapil and Angelos Michaelides and Christoph Schran},
url = {https://www.pnas.org/doi/full/10.1073/pnas.2502684122},
doi = {10.1073/pnas.2502684122},
year = {2025},
date = {2025-08-20},
urldate = {2025-08-20},
journal = {Proc. Natl. Acad. Sci.},
volume = {122},
number = {34},
pages = {e2502684122},
abstract = {An understanding of the $CO_2 + H_2O$ hydration reaction is crucial for modeling the effects of ocean acidification, for enabling novel carbon storage solutions, and as a model process in the geosciences. While the mechanism of this reaction has been investigated extensively in the condensed phase, its mechanism at the air–water interface remains elusive, leaving uncertain the contribution that surface-adsorbed $CO_2$ makes to the overall acidification reaction. In this study, we employ machine-learned potentials trained to various levels of theory to provide a molecular-level understanding of $CO_2$ hydration at the air–water interface. We show that reaction at the interface follows a surface-mediated “in-and-out” mechanism: $CO_2$ diffuses into the aqueous surface layer, reacts to form carbonic acid, and is subsequently expelled from solution. We show that this surface layer provides a bulk-like solvation environment, engendering similar modes of reactivity and near-identical free energy profiles for the bulk and interfacial processes. Our study unveils an unconventional reaction mechanism that underscores the dynamic nature of the molecular reaction site at the air–water interface. The similarity between bulk and interfacial profiles shows that $CO_2$ hydration is equally as feasible under these two solvation environments and that acidification rates are likely enhanced by this additional surface contribution.},
keywords = {Machine Learning Potentials, Water at Interfaces},
pubstate = {published},
tppubtype = {article}
}

Yongkang Wang, Haojian Luo, Xavier R. Advincula, Zhengpu Zhao, Ali Esfandiar, Da Wu, Kara D. Fong, Lei Gao, Arsh S. Hazrah, Takashi Taniguchi, Christoph Schran, Yuki Nagata, Lydéric Bocquet, Marie-Laure Bocquet, Ying Jiang, Angelos Michaelides, Mischa Bonn
Spontaneous surface charging and Janus nature of the hexagonal boron nitride–water interface Journal Article
In: J. Am. Chem. Soc., vol. 147, iss. 33, pp. 30107–30116, 2025.
Abstract | Links | BibTeX | Tags: Charge transfer, Machine Learning Potentials, Water at Interfaces
@article{Wang2025/10.1021/jacs.5c07827,
title = {Spontaneous surface charging and Janus nature of the hexagonal boron nitride–water interface},
author = {Yongkang Wang and Haojian Luo and Xavier R. Advincula and Zhengpu Zhao and Ali Esfandiar and Da Wu and Kara D. Fong and Lei Gao and Arsh S. Hazrah and Takashi Taniguchi and Christoph Schran and Yuki Nagata and Lydéric Bocquet and Marie-Laure Bocquet and Ying Jiang and Angelos Michaelides and Mischa Bonn},
url = {https://pubs.acs.org/doi/full/10.1021/jacs.5c07827},
doi = {10.1021/jacs.5c07827},
year = {2025},
date = {2025-08-06},
urldate = {2025-08-06},
journal = {J. Am. Chem. Soc.},
volume = {147},
issue = {33},
pages = {30107–30116},
abstract = {Boron, nitrogen, and carbon are neighbors in the periodic table and can form strikingly similar twin structures─hexagonal boron nitride (hBN) and graphene─yet nanofluidic experiments demonstrate drastically different water friction on them. We investigate this discrepancy by probing the interfacial water and atomic-scale properties of hBN using surface-specific vibrational spectroscopy, atomic-resolution atomic force microscopy (AFM), and machine learning-based molecular dynamics. Spectroscopy reveals that pristine hBN acquires significant negative charges upon contacting water at neutral pH, unlike hydrophobic graphene, leading to interfacial water alignment and stronger hydrogen bonding. AFM supports that this charging is not defect-induced. pH-dependent measurements suggest $OH^–$ chemisorption and physisorption, which simulations validate as two nearly equally stable states undergoing dynamic exchange. These findings challenge the notion of hBN as chemically inert and hydrophobic, revealing its spontaneous surface charging and Janus nature, and providing molecular insights into its higher water friction compared to carbon surfaces.},
keywords = {Charge transfer, Machine Learning Potentials, Water at Interfaces},
pubstate = {published},
tppubtype = {article}
}
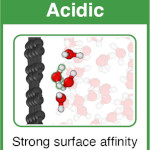
Xavier R. Advincula, Kara D. Fong, Angelos Michaelides, Christoph Schran
Protons accumulate at the graphene–water interface Journal Article
In: ACS Nano, vol. 19, iss. 18, pp. 17728–17737, 2025.
Abstract | Links | BibTeX | Tags: Hydrogen bonding, Machine Learning Potentials, Water, Water at Interfaces
@article{Advincula2025/10.1021/acsnano.5c02053,
title = {Protons accumulate at the graphene–water interface},
author = {Xavier R. Advincula and Kara D. Fong and Angelos Michaelides and Christoph Schran},
url = {https://pubs.acs.org/doi/full/10.1021/acsnano.5c02053},
doi = {10.1021/acsnano.5c02053},
year = {2025},
date = {2025-04-28},
urldate = {2025-04-28},
journal = {ACS Nano},
volume = {19},
issue = {18},
pages = {17728–17737},
abstract = {Water’s ability to autoionize into hydroxide and hydronium ions profoundly influences surface properties, rendering interfaces either basic or acidic. While it is well-established that protons show an affinity to the air–water interface, a critical knowledge gap exists in technologically relevant surfaces like the graphene–water interface. Here we use machine learning-based simulations with first-principles accuracy to unravel the behavior of hydroxide and hydronium ions at the graphene–water interface. Our findings reveal that protons accumulate at the graphene–water interface, with the hydronium ion predominantly residing in the first contact layer of water. In contrast, the hydroxide ion exhibits a bimodal distribution, found both near the surface and further away from it. Analysis of the underlying electronic structure reveals local polarization effects, resulting in counterintuitive charge rearrangement. Proton propensity to the graphene–water interface challenges the interpretation of surface experiments and is expected to have far-reaching consequences for ion conductivity, interfacial reactivity, and proton-mediated processes.},
keywords = {Hydrogen bonding, Machine Learning Potentials, Water, Water at Interfaces},
pubstate = {published},
tppubtype = {article}
}
2024

Fabian L. Thiemann, Niamh O’Neill, Venkat Kapil, Angelos Michaelides, Christoph Schran
Introduction to machine learning potentials for atomistic simulations Journal Article
In: J. Phys.: Condens. Matter, vol. 37, no. 7, pp. 073002, 2024.
Abstract | Links | BibTeX | Tags: Machine Learning Potentials, Potential Energy Surface
@article{Thiemann2024/10.1088/1361-648X/ad9657,
title = {Introduction to machine learning potentials for atomistic simulations},
author = {Fabian L. Thiemann and Niamh O’Neill and Venkat Kapil and Angelos Michaelides and Christoph Schran},
url = {https://iopscience.iop.org/article/10.1088/1361-648X/ad9657/meta},
doi = {10.1088/1361-648X/ad9657},
year = {2024},
date = {2024-12-06},
journal = {J. Phys.: Condens. Matter},
volume = {37},
number = {7},
pages = {073002},
abstract = {Machine learning potentials have revolutionised the field of atomistic simulations in recent years and are becoming a mainstay in the toolbox of computational scientists. This paper aims to provide an overview and introduction into machine learning potentials and their practical application to scientific problems. We provide a systematic guide for developing machine learning potentials, reviewing chemical descriptors, regression models, data generation and validation approaches. We begin with an emphasis on the earlier generation of models, such as high-dimensional neural network potentials and Gaussian approximation potentials, to provide historical perspective and guide the reader towards the understanding of recent developments, which are discussed in detail thereafter. Furthermore, we refer to relevant expert reviews, open-source software, and practical examples—further lowering the barrier to exploring these methods. The paper ends with selected showcase examples, highlighting the capabilities of machine learning potentials and how they can be applied to push the boundaries in atomistic simulations.},
keywords = {Machine Learning Potentials, Potential Energy Surface},
pubstate = {published},
tppubtype = {article}
}

Niamh O'Neill, Christoph Schran, Stephen J. Cox, Angelos Michaelides
Crumbling crystals: on the dissolution mechanism of NaCl in water Journal Article
In: Phys. Chem. Chem. Phys., vol. 26, iss. 42, pp. 26933-26942, 2024.
Abstract | Links | BibTeX | Tags: Ions in Water, Machine Learning Potentials, Water
@article{ONeill2024/10.1039/d4cp03115f,
title = {Crumbling crystals: on the dissolution mechanism of NaCl in water},
author = {Niamh O'Neill and Christoph Schran and Stephen J. Cox and Angelos Michaelides},
url = {https://pubs.rsc.org/en/content/articlelanding/2024/cp/d4cp03115f},
doi = {10.1039/d4cp03115f},
year = {2024},
date = {2024-10-30},
urldate = {2024-10-30},
journal = {Phys. Chem. Chem. Phys.},
volume = {26},
issue = {42},
pages = {26933-26942},
abstract = {Dissolution of ionic salts in water is ubiquitous, particularly for NaCl. However, an atomistic scale understanding of the process remains elusive. Simulations lend themselves conveniently to studying dissolution since they provide the spatio-temporal resolution that can be difficult to obtain experimentally. Nevertheless, the complexity of various inter- and intra-molecular interactions require careful treatment and long time scale simulations, both of which are typically hindered by computational expense. Here, we use advances in machine learning potential methodology to resolve at an ab initio level of theory the dissolution mechanism of NaCl in water. The picture that emerges is that of a steady ion-wise unwrapping of the crystal preceding its rapid disintegration, reminiscent of crumbling. The onset of crumbling can be explained by a strong increase in the ratio of the surface area to volume of the crystal. Overall, dissolution comprises a series of highly dynamical microscopic sub-processes, resulting in an inherently stochastic mechanism. These atomistic level insights contribute to the general understanding of dissolution mechanisms in other crystals, and the methodology is primed for more complex systems of recent interest such as water/salt interfaces under flow and salt crystals under confinement.},
keywords = {Ions in Water, Machine Learning Potentials, Water},
pubstate = {published},
tppubtype = {article}
}

Samuel G. H. Brookes, Venkat Kapil, Christoph Schran, Angelos Michaelides
The wetting of H$_2$O by CO$_2$ Journal Article
In: J. Chem. Phys., vol. 161, no. 8, pp. 084711, 2024, ISSN: 10897690.
Abstract | Links | BibTeX | Tags: Machine Learning Potentials, Water, Water at Interfaces
@article{Brookes2024/10.1063/5.0224230,
title = {The wetting of H$_2$O by CO$_2$},
author = {Samuel G. H. Brookes and Venkat Kapil and Christoph Schran and Angelos Michaelides},
url = {/aip/jcp/article/161/8/084711/3309975/The-wetting-of-H2O-by-CO2},
doi = {10.1063/5.0224230},
issn = {10897690},
year = {2024},
date = {2024-08-28},
urldate = {2024-08-28},
journal = {J. Chem. Phys.},
volume = {161},
number = {8},
pages = {084711},
publisher = {American Institute of Physics},
abstract = {Biphasic interfaces are complex but fascinating regimes that display a number of properties distinct from those of the bulk. The CO2-H2O interface, in particular, has been the subject of a number of studies on account of its importance for the carbon life cycle as well as carbon capture and sequestration schemes. Despite this attention, there remain a number of open questions on the nature of the CO2-H2O interface, particularly concerning the interfacial tension and phase behavior of CO2 at the interface. In this paper, we seek to address these ambiguities using ab initio-quality simulations. Harnessing the benefits of machine-learned potentials and enhanced statistical sampling methods, we present an ab initio-level description of the CO2-H2O interface. Interfacial tensions are predicted from 1 to 500 bars and found to be in close agreement with experiment at pressures for which experimental data are available. Structural analyses indicate the buildup of an adsorbed, saturated CO2 film forming at a low pressure (20 bars) with properties similar to those of the bulk liquid, but preferential perpendicular alignment with respect to the interface. The CO2 monolayer buildup coincides with a reduced structuring of water molecules close to the interface. This study highlights the predictive nature of machine-learned potentials for complex macroscopic properties of biphasic interfaces, and the mechanistic insight obtained into carbon dioxide aggregation at the water interface is of high relevance for geoscience, climate research, and materials science.},
keywords = {Machine Learning Potentials, Water, Water at Interfaces},
pubstate = {published},
tppubtype = {article}
}
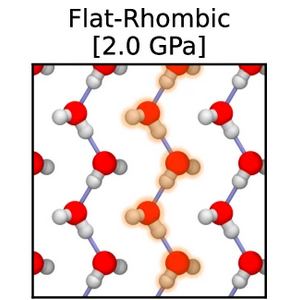
Pavan Ravindra, Xavier R. Advincula, Christoph Schran, Angelos Michaelides, Venkat Kapil
Quasi-one-dimensional hydrogen bonding in nanoconfined ice Journal Article
In: Nat. Commun., vol. 15, no. 1, pp. 1–9, 2024, ISSN: 20411723.
Abstract | Links | BibTeX | Tags: Confinement, Hydrogen bonding, Machine Learning Potentials, Water
@article{Ravindra2024/10.1038/s41467-024-51124-z,
title = {Quasi-one-dimensional hydrogen bonding in nanoconfined ice},
author = {Pavan Ravindra and Xavier R. Advincula and Christoph Schran and Angelos Michaelides and Venkat Kapil},
url = {https://www.nature.com/articles/s41467-024-51124-z},
doi = {10.1038/s41467-024-51124-z},
issn = {20411723},
year = {2024},
date = {2024-08-24},
urldate = {2024-08-24},
journal = {Nat. Commun.},
volume = {15},
number = {1},
pages = {1–9},
publisher = {Nature Publishing Group},
abstract = {The Bernal-Fowler ice rules stipulate that each water molecule in an ice crystal should form four hydrogen bonds. However, in extreme or constrained conditions, the arrangement of water molecules deviates from conventional ice rules, resulting in properties significantly different from bulk water. In this study, we employ machine learning-driven first-principles simulations to identify a new stabilization mechanism in nanoconfined ice phases. Instead of forming four hydrogen bonds, nanoconfined crystalline ice can form a quasi-one-dimensional hydrogen-bonded structure that exhibits only two hydrogen bonds per water molecule. These structures consist of strongly hydrogen-bonded linear chains of water molecules that zig-zag along one dimension, stabilized by van der Waals interactions that stack these chains along the other dimension. The unusual interplay of hydrogen bonding and van der Waals interactions in nanoconfined ice results in atypical proton behavior such as potential ferroelectric behavior, low dielectric response, and long-range proton dynamics.},
keywords = {Confinement, Hydrogen bonding, Machine Learning Potentials, Water},
pubstate = {published},
tppubtype = {article}
}
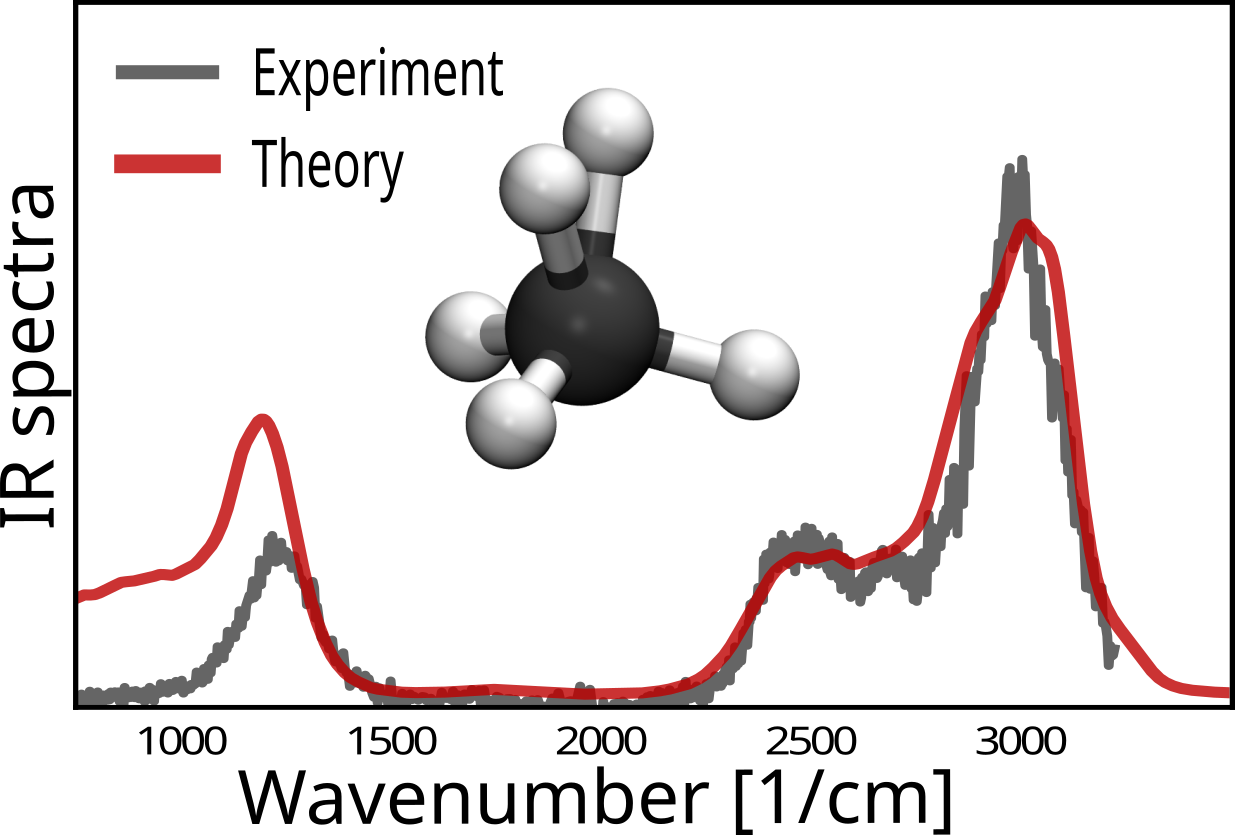
Richard Beckmann, Christoph Schran, Fabien Brieuc, Dominik Marx
Theoretical infrared spectroscopy of protonated methane isotopologues Journal Article
In: Phys. Chem. Chem. Phys., vol. 26, iss. 35, pp. 22846-22852, 2024.
Abstract | Links | BibTeX | Tags: Coupled Cluster, Quantum Dynamics, Spectra
@article{Beckmann2024/10.1039/D4CP02295E,
title = {Theoretical infrared spectroscopy of protonated methane isotopologues},
author = {Richard Beckmann and Christoph Schran and Fabien Brieuc and Dominik Marx},
url = {http://dx.doi.org/10.1039/D4CP02295E},
doi = {10.1039/D4CP02295E},
year = {2024},
date = {2024-08-13},
urldate = {2024-08-13},
journal = {Phys. Chem. Chem. Phys.},
volume = {26},
issue = {35},
pages = {22846-22852},
publisher = {The Royal Society of Chemistry},
abstract = {The vibrational spectroscopy of protonated methane and its mixed hydrogen/deuterium isotopologues remains a challenge to both experimental and computational spectroscopy due to the iconic floppiness of CH5+. Here, we compute the finite-temperature broadband infrared spectra of CH5+ and all its isotopologues, i.e. CHnD5−n+ up to CD5+, from path integral molecular dynamics in conjunction with interactions and dipoles computed consistently at CCSD(T) coupled cluster accuracy. The potential energy and dipole moment surfaces have been accurately represented in full dimensionality in terms of high-dimensional neural networks. The resulting computational efficiency allows us to establish CCSD(T) accuracy at the level of converged path integral simulations. For all six isotopologues, the computed broadband spectra compare very favorably to the available experimental broadband spectra obtained from laser induced reactions action vibrational spectroscopy. The current approach is found to consistently and significantly improve on previous calculations of these broadband vibrational spectra and defines the new cutting-edge for what has been dubbed the “enfant terrible” of molecular spectroscopy in view of its pronounced large-amplitude motion that involves all intramolecular degrees of freedom.},
keywords = {Coupled Cluster, Quantum Dynamics, Spectra},
pubstate = {published},
tppubtype = {article}
}
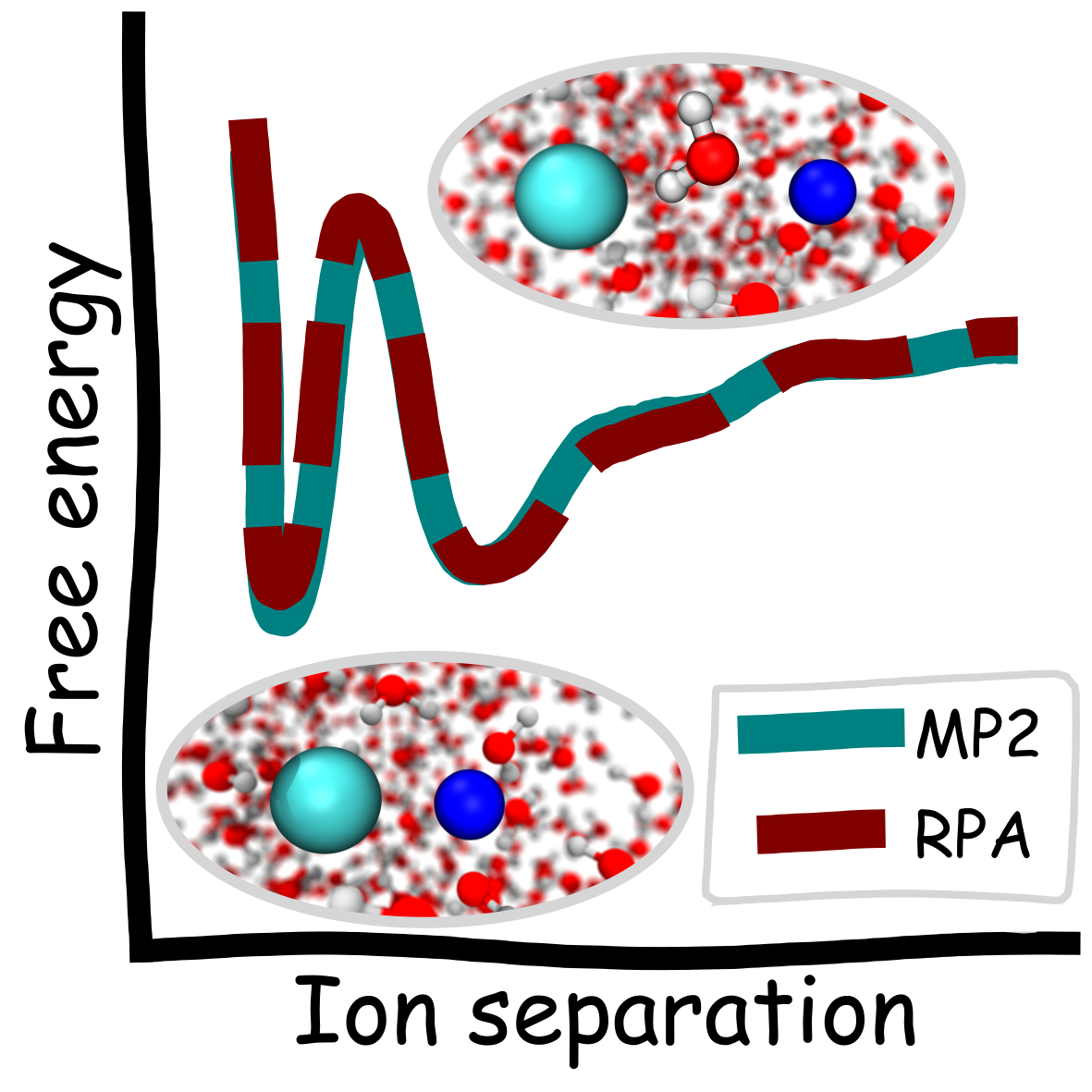
Niamh O’Neill, Benjamin X. Shi, Kara D. Fong, Angelos Michaelides, Christoph Schran
To pair or not to pair? Machine-learned explicitly-correlated electronic structure for NaCl in water Journal Article
In: The Journal of Physical Chemistry Letters, vol. 15, no. 23, pp. 6081–6091, 2024, (PMID: 38820256).
Abstract | Links | BibTeX | Tags: Ions in Water, Machine Learning Potentials
@article{ONeill/10.1021/acs.jpclett.4c01030,
title = {To pair or not to pair? Machine-learned explicitly-correlated electronic structure for NaCl in water},
author = {Niamh O’Neill and Benjamin X. Shi and Kara D. Fong and Angelos Michaelides and Christoph Schran},
url = {https://doi.org/10.1021/acs.jpclett.4c01030},
doi = {10.1021/acs.jpclett.4c01030},
year = {2024},
date = {2024-05-31},
urldate = {2024-05-31},
journal = {The Journal of Physical Chemistry Letters},
volume = {15},
number = {23},
pages = {6081–6091},
abstract = {The extent of ion pairing in solution is an important phenomenon to rationalize transport and thermodynamic properties of electrolytes. A fundamental measure of this pairing is the potential of mean force (PMF) between solvated ions. The relative stabilities of the paired and solvent shared states in the PMF and the barrier between them are highly sensitive to the underlying potential energy surface. However, direct application of accurate electronic structure methods is challenging, since long simulations are required. We develop wave function based machine learning potentials with the random phase approximation (RPA) and second order Møller–Plesset (MP2) perturbation theory for the prototypical system of Na and Cl ions in water. We show both methods in agreement, predicting the paired and solvent shared states to have similar energies (within 0.2 kcal/mol). We also provide the same benchmarks for different DFT functionals as well as insight into the PMF based on simple analyses of the interactions in the system.},
note = {PMID: 38820256},
keywords = {Ions in Water, Machine Learning Potentials},
pubstate = {published},
tppubtype = {article}
}
Kara D. Fong, Barbara Sumić, Niamh O’Neill, Christoph Schran, Clare P. Grey, Angelos Michaelides
The interplay of solvation and polarization effects on ion pairing in nanoconfined electrolytes Journal Article
In: Nano Letters, vol. 24, no. 16, pp. 5024–5030, 2024, (PMID: 38592099).
Abstract | Links | BibTeX | Tags: Confinement, Hydrogen bonding, Ions in Water, Machine Learning Potentials
@article{Fong/10.1021/acs.nanolett.4c00890,
title = {The interplay of solvation and polarization effects on ion pairing in nanoconfined electrolytes},
author = {Kara D. Fong and Barbara Sumić and Niamh O’Neill and Christoph Schran and Clare P. Grey and Angelos Michaelides},
url = {https://doi.org/10.1021/acs.nanolett.4c00890},
doi = {10.1021/acs.nanolett.4c00890},
year = {2024},
date = {2024-04-09},
urldate = {2024-04-09},
journal = {Nano Letters},
volume = {24},
number = {16},
pages = {5024–5030},
abstract = {The nature of ion–ion interactions in electrolytes confined to nanoscale pores has important implications for energy storage and separation technologies. However, the physical effects dictating the structure of nanoconfined electrolytes remain debated. Here we employ machine-learning-based molecular dynamics simulations to investigate ion–ion interactions with density functional theory level accuracy in a prototypical confined electrolyte, aqueous NaCl within graphene slit pores. We find that the free energy of ion pairing in highly confined electrolytes deviates substantially from that in bulk solutions, observing a decrease in contact ion pairing but an increase in solvent-separated ion pairing. These changes arise from an interplay of ion solvation effects and graphene’s electronic structure. Notably, the behavior observed from our first-principles-level simulations is not reproduced even qualitatively with the classical force fields conventionally used to model these systems. The insight provided in this work opens new avenues for predicting and controlling the structure of nanoconfined electrolytes.},
note = {PMID: 38592099},
keywords = {Confinement, Hydrogen bonding, Ions in Water, Machine Learning Potentials},
pubstate = {published},
tppubtype = {article}
}
Thomas Dufils, Christoph Schran, Ji Chen, Andre K. Geim, Laura Fumagalli, Angelos Michaelides
Origin of dielectric polarization suppression in confined water from first principles Journal Article
In: Chem. Sci., vol. 15, iss. 2, pp. 516–527, 2024.
Abstract | Links | BibTeX | Tags: AIMD, Confinement, Hydrogen bonding, Water
@article{Dufils2024/10.1039/D3SC04740G,
title = {Origin of dielectric polarization suppression in confined water from first principles},
author = {Thomas Dufils and Christoph Schran and Ji Chen and Andre K. Geim and Laura Fumagalli and Angelos Michaelides},
url = {http://dx.doi.org/10.1039/D3SC04740G},
doi = {10.1039/D3SC04740G},
year = {2024},
date = {2024-01-01},
urldate = {2024-01-01},
journal = {Chem. Sci.},
volume = {15},
issue = {2},
pages = {516–527},
publisher = {The Royal Society of Chemistry},
abstract = {It has long been known that the dielectric constant of confined water should be different from that in bulk. Recent experiments have shown that it is vanishingly small, however the origin of the phenomenon remains unclear. Here we used ab initio molecular dynamics simulations (AIMD) and AIMD-trained machine-learning potentials to understand water's structure and electronic properties underpinning this effect. For the graphene and hexagonal boron-nitride substrates considered, we find that it originates in the spontaneous anti-parallel alignment of the water dipoles in the first two water layers near the solid interface. The interfacial layers exhibit net ferroelectric ordering, resulting in an overall anti-ferroelectric arrangement of confined water. Together with constrained hydrogen-bonding orientations, this leads to much reduced out-of-plane polarization. Furthermore, we directly contrast AIMD and simple classical force-field simulations, revealing important differences. This work offers insight into a property of water that is critical in modulating surface forces, the electric-double-layer formation and molecular solvation, and shows a way to compute it.},
keywords = {AIMD, Confinement, Hydrogen bonding, Water},
pubstate = {published},
tppubtype = {article}
}
2023
Fabien Brieuc, Christoph Schran, Dominik Marx
Manifestations of local supersolidity of $^4$He around a charged molecular impurity Journal Article
In: Phys. Rev. Res., vol. 5, iss. 4, pp. 043083, 2023.
Links | BibTeX | Tags: Nuclear quantum effects, path integral molecular dynamics (PIMD), Superfluidity
@article{PhysRevResearch.5.043083,
title = {Manifestations of local supersolidity of $^4$He around a charged molecular impurity},
author = {Fabien Brieuc and Christoph Schran and Dominik Marx},
url = {https://link.aps.org/doi/10.1103/PhysRevResearch.5.043083},
doi = {10.1103/PhysRevResearch.5.043083},
year = {2023},
date = {2023-10-01},
urldate = {2023-10-01},
journal = {Phys. Rev. Res.},
volume = {5},
issue = {4},
pages = {043083},
publisher = {American Physical Society},
keywords = {Nuclear quantum effects, path integral molecular dynamics (PIMD), Superfluidity},
pubstate = {published},
tppubtype = {article}
}
Julia A. Davies, Christoph Schran, Fabien Brieuc, Dominik Marx, Andrew M. Ellis
Onset of rotational decoupling for a molecular ion solvated in helium: From tags to rings and shells Journal Article
In: Phys. Rev. Lett., vol. 130, iss. 8, pp. 083001, 2023.
Links | BibTeX | Tags: Nuclear quantum effects, path integral molecular dynamics (PIMD), Superfluidity, Water
@article{Schran2023/10.1103/PhysRevLett.130.083001,
title = {Onset of rotational decoupling for a molecular ion solvated in helium: From tags to rings and shells},
author = {Julia A. Davies and Christoph Schran and Fabien Brieuc and Dominik Marx and Andrew M. Ellis},
doi = {10.1103/PhysRevLett.130.083001},
year = {2023},
date = {2023-02-01},
urldate = {2023-02-01},
journal = {Phys. Rev. Lett.},
volume = {130},
issue = {8},
pages = {083001},
publisher = {American Physical Society},
keywords = {Nuclear quantum effects, path integral molecular dynamics (PIMD), Superfluidity, Water},
pubstate = {published},
tppubtype = {article}
}
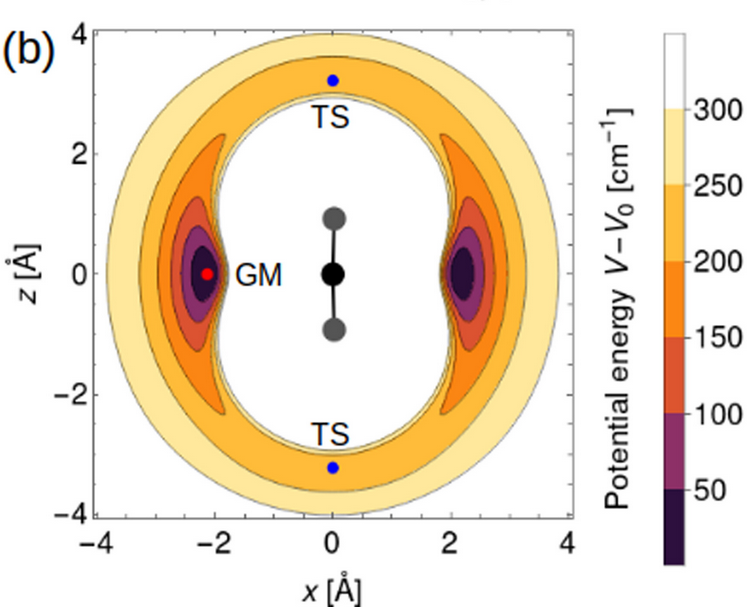
Irén Simkó, Christoph Schran, Fabien Brieuc, Csaba Fábri, Oskar Asvany, Stephan Schlemmer, Dominik Marx, Attila G. Császár
Quantum nuclear delocalization and its rovibrational fingerprints Journal Article
In: Angewandte Chemie International Edition, vol. 62, no. 41, pp. e202306744, 2023.
Abstract | Links | BibTeX | Tags: Nuclear quantum effects, path integral molecular dynamics (PIMD), Quantum Dynamics
@article{Simko/10.1002/anie.202306744,
title = {Quantum nuclear delocalization and its rovibrational fingerprints},
author = {Irén Simkó and Christoph Schran and Fabien Brieuc and Csaba Fábri and Oskar Asvany and Stephan Schlemmer and Dominik Marx and Attila G. Császár},
url = {https://onlinelibrary.wiley.com/doi/abs/10.1002/anie.202306744},
doi = {10.1002/anie.202306744},
year = {2023},
date = {2023-01-01},
urldate = {2023-01-01},
journal = {Angewandte Chemie International Edition},
volume = {62},
number = {41},
pages = {e202306744},
abstract = {Abstract Quantum mechanics dictates that nuclei must undergo some delocalization. In this work, emergence of quantum nuclear delocalization and its rovibrational fingerprints are discussed for the case of the van der Waals complex . The equilibrium structure of is planar and T-shaped, one He atom solvating the quasi-linear He−H+−He core. The dynamical structure of , in all of its bound states, is fundamentally different. As revealed by spatial distribution functions and nuclear densities, during the vibrations of the molecule the solvating He is not restricted to be in the plane defined by the instantaneously bent chomophore, but freely orbits the central proton, forming a three-dimensional torus around the chromophore. This quantum delocalization is observed for all vibrational states, the type of vibrational excitation being reflected in the topology of the nodal surfaces in the nuclear densities, showing, for example, that intramolecular bending involves excitation along the circumference of the torus.},
keywords = {Nuclear quantum effects, path integral molecular dynamics (PIMD), Quantum Dynamics},
pubstate = {published},
tppubtype = {article}
}
2022

Venkat Kapil, Christoph Schran, Andrea Zen, Ji Chen, Chris J. Pickard, Angelos Michaelides
The first-principles phase diagram of monolayer nanoconfined water Journal Article
In: Nature, vol. 609, pp. 512-516, 2022, ISSN: 1476-4687.
Abstract | Links | BibTeX | Tags: Confinement, Machine Learning Potentials, Water
@article{Kapil2022/10.1038/S41586-022-05036-X,
title = {The first-principles phase diagram of monolayer nanoconfined water},
author = {Venkat Kapil and Christoph Schran and Andrea Zen and Ji Chen and Chris J. Pickard and Angelos Michaelides},
doi = {10.1038/S41586-022-05036-X},
issn = {1476-4687},
year = {2022},
date = {2022-09-01},
urldate = {2022-09-01},
journal = {Nature},
volume = {609},
pages = {512-516},
publisher = {Nature Publishing Group},
abstract = {Water in nanoscale cavities is ubiquitous and of central importance to everyday phenomena in geology and biology. However, the properties of nanoscale water can be substantially different from those of bulk water, as shown, for example, by the anomalously low dielectric constant of water in nanochannels1, near frictionless water flow2 or the possible existence of a square ice phase3. Such properties suggest that nanoconfined water could be engineered for technological applications in nanofluidics4, electrolyte materials5 and water desalination6. Unfortunately, challenges in experimentally characterizing water at the nanoscale and the high cost of first-principles simulations have prevented the molecular-level understanding required to control the behaviour of water. Here we combine a range of computational approaches to enable a first-principles-level investigation of a single layer of water within a graphene-like channel. We find that monolayer water exhibits surprisingly rich and diverse phase behaviour that is highly sensitive to temperature and the van der Waals pressure acting within the nanochannel. In addition to multiple molecular phases with melting temperatures varying non-monotonically by more than 400 kelvins with pressure, we predict a hexatic phase, which is an intermediate between a solid and a liquid, and a superionic phase with a high electrical conductivity exceeding that of battery materials. Notably, this suggests that nanoconfinement could be a promising route towards superionic behaviour under easily accessible conditions. Monolayer water exhibits rich and diverse phase behaviour that is highly sensitive to temperature and the van der Waals pressure acting within the nanochannel.},
keywords = {Confinement, Machine Learning Potentials, Water},
pubstate = {published},
tppubtype = {article}
}
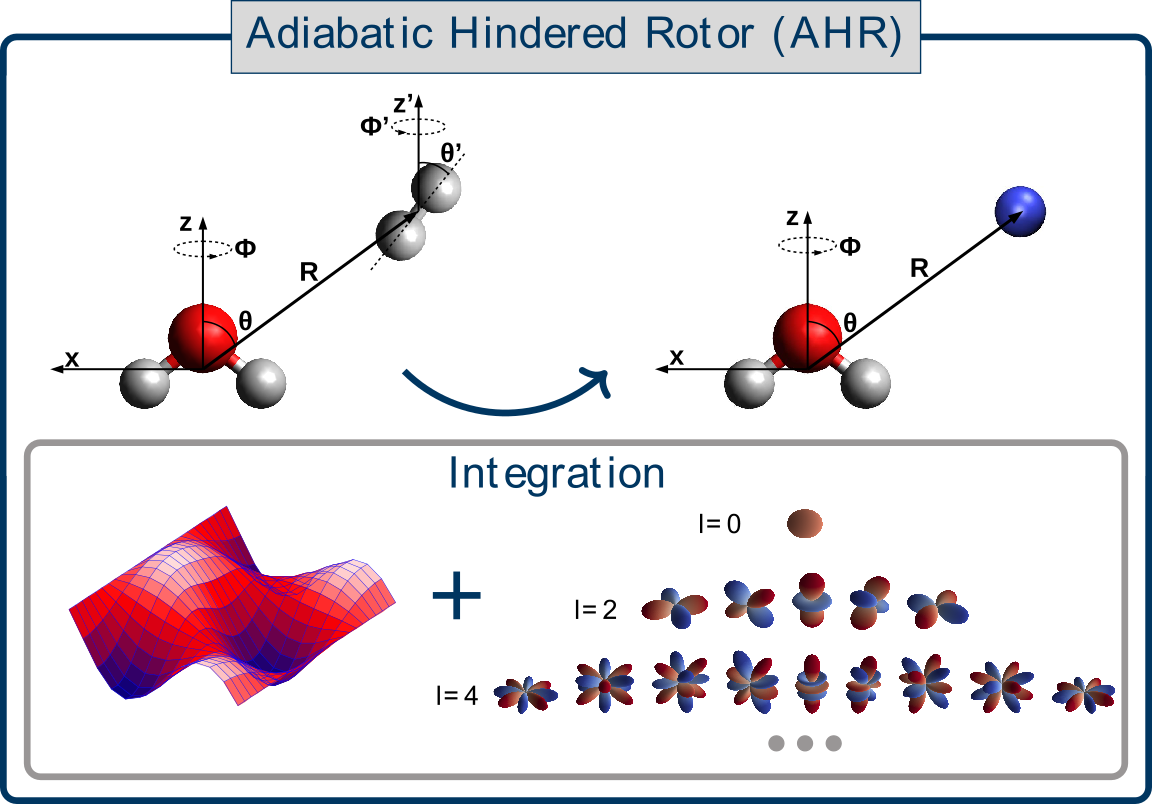
Laura Durán Caballero, Christoph Schran, Fabien Brieuc, Dominik Marx
Neural network interaction potentials for para-hydrogen with flexible molecules Journal Article
In: J. Chem. Phys., vol. 157, no. 7, pp. 074302, 2022, ISSN: 0021-9606.
Abstract | Links | BibTeX | Tags: Machine Learning Potentials, Nuclear quantum effects, Superfluidity, Water
@article{Duran2022/10.1063/5.0100953,
title = {Neural network interaction potentials for para-hydrogen with flexible molecules},
author = {Laura Durán Caballero and Christoph Schran and Fabien Brieuc and Dominik Marx},
doi = {10.1063/5.0100953},
issn = {0021-9606},
year = {2022},
date = {2022-08-01},
urldate = {2022-08-01},
journal = {J. Chem. Phys.},
volume = {157},
number = {7},
pages = {074302},
publisher = {AIP Publishing LLCAIP Publishing},
abstract = {The study of molecular impurities in para-hydrogen (pH2) clusters is key to push forward our understanding of intra- and intermolecular interactions including their impact on the superfluid response of this bosonic quantum solvent. This includes tagging with one or very few pH2, the microsolvation regime, and matrix isolation. However, the fundamental coupling between the bosonic pH2 environment and the (ro-)vibrational motion of molecular impurities remains poorly understood. Quantum simulations can in provide the necessary atomistic insight, but very accurate descriptions of the involved interactions are required. Here, we present a data-driven approach for the generation of impurity-pH2 interaction potentials based on machine learning techniques which retain the full flexibility of the impurity. We employ the well-established adiabatic hindered rotor (AHR) averaging technique to include the impact of the nuclear spin statistics on the symmetry-allowed rotational quantum numbers of pH2. Embedding this averaging procedure within the high-dimensional neural network potential (NNP) framework enables the generation of highly-accurate AHR-averaged NNPs at coupled cluster accuracy, namely CCSD(T*)-F12a/aVTZcp in an automated manner. We apply this methodology to the water and protonated water molecules, as representative cases for quasi-rigid and highly-flexible molecules respectively, and obtain AHR-averaged NNPs that reliably describe the H2O-pH2 and H3O+-pH2 interactions. Using path integral simulations we show for the hydronium cation that umbrella-like tunneling inversion has a strong impact on the first and second pH2 microsolvation shells. The data-driven nature of our protocol opens the door to the study of bosonic pH2 quantum solvation for a wide range of embedded impurities.},
keywords = {Machine Learning Potentials, Nuclear quantum effects, Superfluidity, Water},
pubstate = {published},
tppubtype = {article}
}
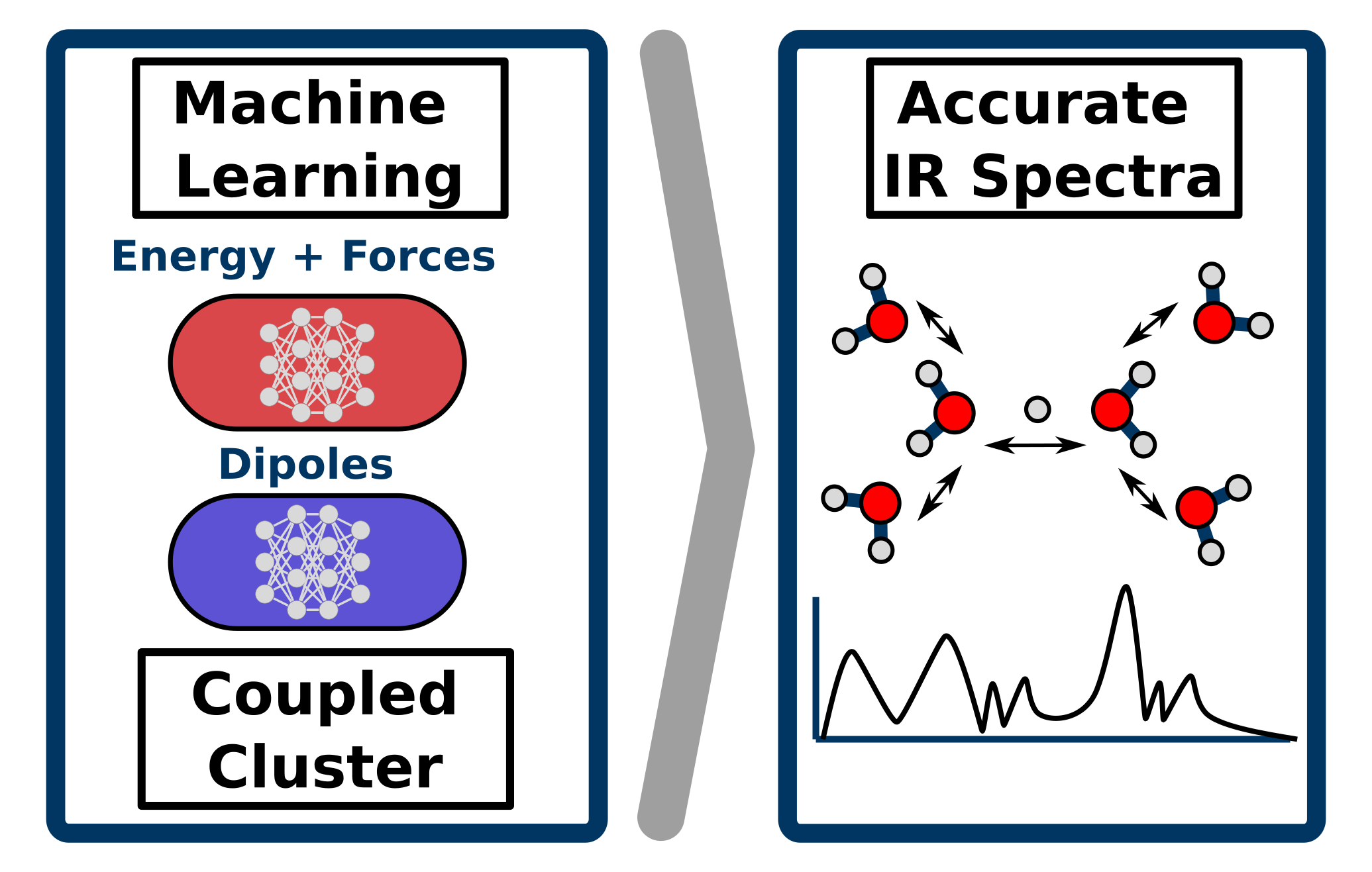
Richard Beckmann, Fabien Brieuc, Christoph Schran, Dominik Marx
Infrared spectra at coupled cluster accuracy from neural network representations Journal Article
In: J. Chem. Theory Comput., 2022, ISSN: 1549-9618.
Abstract | Links | BibTeX | Tags: Coupled Cluster, Machine Learning Potentials, Spectra, Water
@article{Beckmann2022/10.1021/ACS.JCTC.2C00511,
title = {Infrared spectra at coupled cluster accuracy from neural network representations},
author = {Richard Beckmann and Fabien Brieuc and Christoph Schran and Dominik Marx},
doi = {10.1021/ACS.JCTC.2C00511},
issn = {1549-9618},
year = {2022},
date = {2022-08-01},
urldate = {2022-08-01},
journal = {J. Chem. Theory Comput.},
publisher = {American Chemical Society},
abstract = {Infrared spectroscopy is key to elucidate molecular structures, monitor reactions and observe conformational changes, while providing information on both structural and dynamical properties. This makes the accurate prediction of infrared spectra based on first-principle theories a highly desirable pursuit. Molecular dynamics simulations have proven to be a particularly powerful approach for this task, albeit requiring the computation of energies, forces and dipole moments for a large number of molecular configurations as a function of time. This explains why highly accurate first principles methods, such as coupled cluster theory, have so far been inapplicable for the prediction of fully anharmonic vibrational spectra of large systems at finite temperatures. Here, we push cutting-edge machine learning techniques forward by using neural network representations of energies, forces and in particular dipoles to predict such infrared spectra fully at "gold standard" coupled cluster accuracy as demonstrated for protonated water clusters as large as the protonated water hexamer, in its extended Zundel configuration. Furthermore, we show that this methodology can be used beyond the scope of the data considered during the development of the neural network models, allowing for the computation of finite-temperature infrared spectra of large systems inaccessible to explicit coupled cluster calculations. This substantially expands the hitherto existing limits of accuracy, speed and system size for theoretical spectroscopy and opens up a multitude of avenues for the prediction of vibrational spectra and the understanding of complex intra- and intermolecular couplings.},
keywords = {Coupled Cluster, Machine Learning Potentials, Spectra, Water},
pubstate = {published},
tppubtype = {article}
}
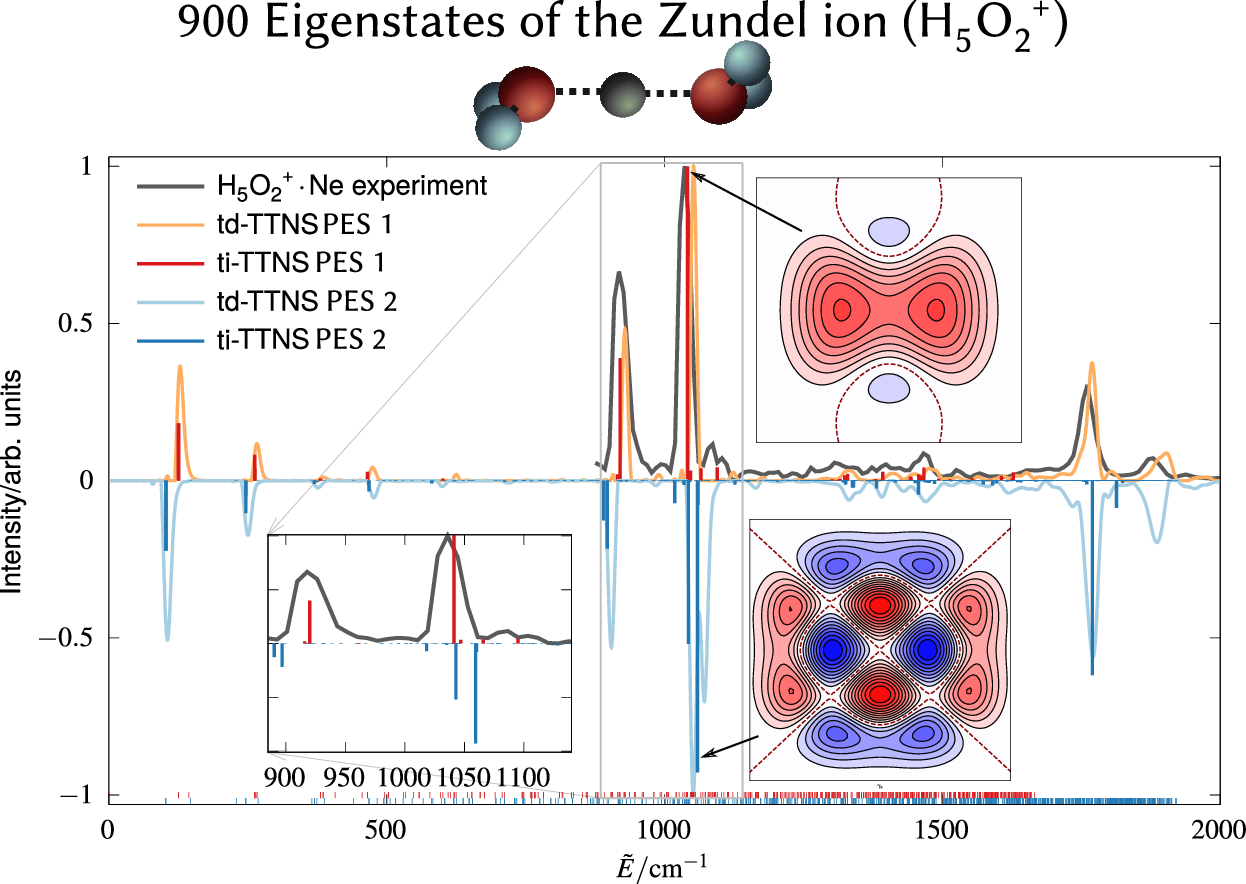
Henrik R Larsson, Markus Schröder, Richard Beckmann, Fabien Brieuc, Christoph Schran, Dominik Marx, Oriol Vendrell
State-resolved infrared spectrum of the protonated water dimer: Revisiting the characteristic proton transfer doublet peak Journal Article
In: Chem. Sci., 2022, ISSN: 2041-6539.
Abstract | Links | BibTeX | Tags: Quantum Dynamics, Spectra, Water
@article{Larsson2022/10.1039/D2SC03189B,
title = {State-resolved infrared spectrum of the protonated water dimer: Revisiting the characteristic proton transfer doublet peak},
author = {Henrik R Larsson and Markus Schröder and Richard Beckmann and Fabien Brieuc and Christoph Schran and Dominik Marx and Oriol Vendrell},
doi = {10.1039/D2SC03189B},
issn = {2041-6539},
year = {2022},
date = {2022-08-01},
urldate = {2022-08-01},
journal = {Chem. Sci.},
publisher = {The Royal Society of Chemistry},
abstract = {The infrared (IR) spectra of protonated water clusters encode precise information on the dynamics and structure of the hydrated proton. However, the strong anharmonic coupling and quantum effects of these elusive species remain puzzling up to the present day. Here, we report unequivocal evidence that the interplay between the proton transfer and the water wagging motions in the protonated water dimer (Zundel ion) giving rise to the characteristic doublet peak is both more complex and more sensitive to subtle energetic changes than previously thought. In particular, hitherto overlooked low-intensity satellite peaks in the experimental spectrum are now unveiled and mechanistically assigned. Our findings rely on the comparison of IR spectra obtained using two highly accurate potential energy surfaces in conjunction with highly accurate state-resolved quantum simulations. We demonstrate that these high-accuracy simulations are important for providing definite assignments of the complex IR signals of fluxional molecules.},
keywords = {Quantum Dynamics, Spectra, Water},
pubstate = {published},
tppubtype = {article}
}
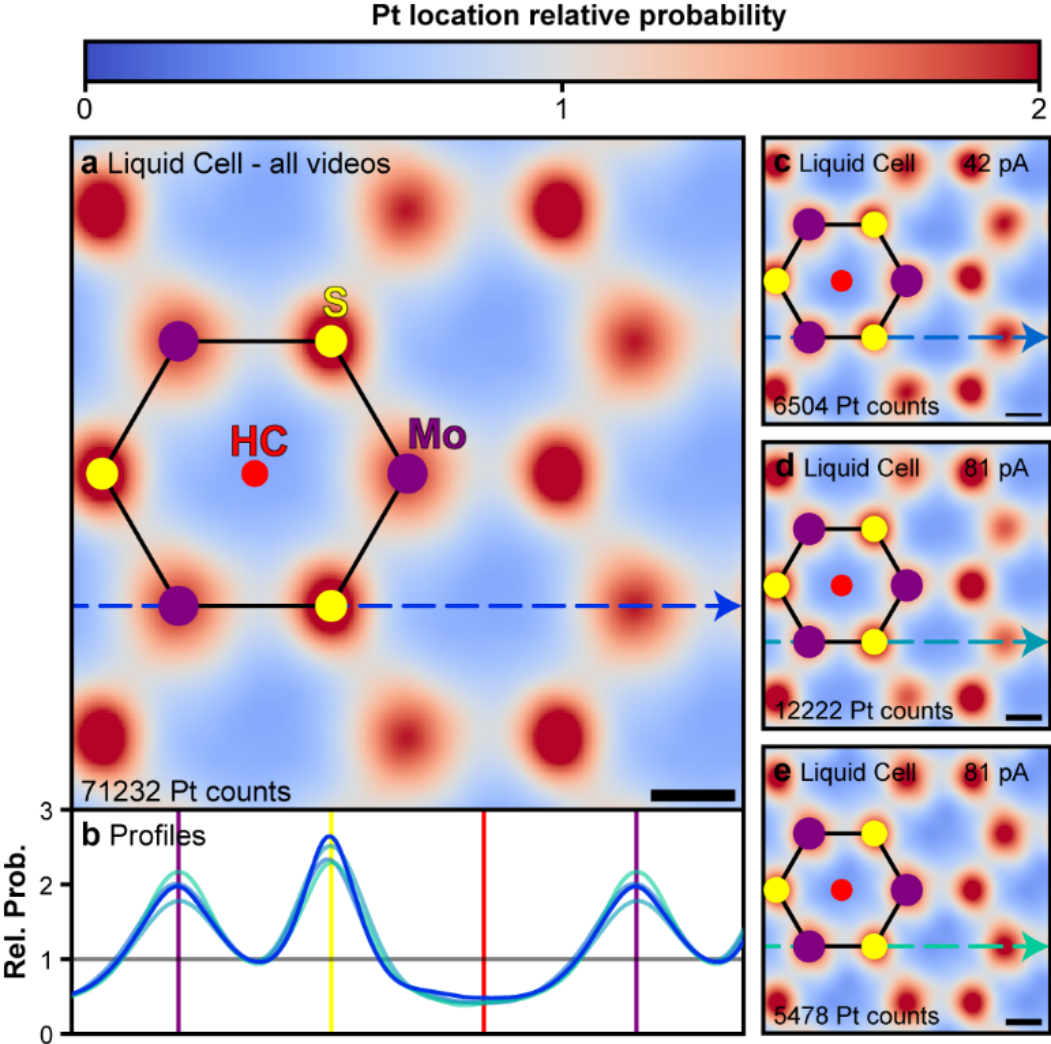
Nick Clark, Daniel J. Kelly, Mingwei Zhou, Yi-Chao Zou, Chang Woo Myung, David G. Hopkinson, Christoph Schran, Angelos Michaelides, Roman Gorbachev, Sarah J. Haigh
Tracking single adatoms in liquid in a Transmission Electron Microscope Journal Article
In: Nature, pp. 1–3, 2022, ISSN: 1476-4687.
Abstract | Links | BibTeX | Tags: Two-dimensional materials, Water at Interfaces
@article{Clark2022/10.1038/s41586-022-05130-0,
title = {Tracking single adatoms in liquid in a Transmission Electron Microscope},
author = {Nick Clark and Daniel J. Kelly and Mingwei Zhou and Yi-Chao Zou and Chang Woo Myung and David G. Hopkinson and Christoph Schran and Angelos Michaelides and Roman Gorbachev and Sarah J. Haigh},
doi = {10.1038/s41586-022-05130-0},
issn = {1476-4687},
year = {2022},
date = {2022-07-01},
urldate = {2022-07-01},
journal = {Nature},
pages = {1–3},
publisher = {Nature Publishing Group},
abstract = {Single atoms or ions on surfaces affect processes from nucleation1 to electrochemical reactions2 and heterogeneous catalysis3. Transmission electron microscopy (TEM) is a leading approach for visualizing single atoms on a variety of substrates4,5. It conventionally requires high vacuum conditions, but has been developed for in situ imaging in liquid and gaseous environments6,7 with a combined spatial and temporal resolution that is unmatched by any other method —notwithstanding concerns about electron beam effects on samples. When imaging in liquid using commercial technologies, electron scattering in the windows enclosing the sample and in the liquid generally limits the achievable resolution to a few nanometres6,8,9. Graphene liquid cells, on the other hand, have enabled atomic resolution imaging of metal nanoparticles in liquids10. Here we show that a double graphene liquid cell, comprised of a central molybdenum disulphide monolayer separated by hexagonal boron nitride spacers from the two enclosing graphene windows, makes it possible to monitor with atomic resolution the dynamics of platinum adatoms on the monolayer in an aqueous salt solution. By imaging over 70,000 single adatom adsorption sites, we compare the site preference and dynamic motion of the adatoms in both a fully hydrated and vacuum state. We find a modified adsorption site distribution and higher diffusivities for the adatoms in liquid phase compared to those in vacuum. This approach paves the way for in situ liquid phase imaging of chemical processes with single atom precision.},
keywords = {Two-dimensional materials, Water at Interfaces},
pubstate = {published},
tppubtype = {article}
}

Fabian L. Thiemann, Christoph Schran, Patrick Rowe, Erich A. Müller, Angelos Michaelides
Water flow in single-wall nanotubes: Oxygen makes it slip, hydrogen makes it stick Journal Article
In: ACS Nano, vol. 16, no. 7, pp. 10775–10782, 2022.
Abstract | Links | BibTeX | Tags: Confinement, Hydrogen bonding, Machine Learning Potentials, Water at Interfaces, Water flow
@article{Thiemann2022/10.1021/acsnano.2c02784,
title = {Water flow in single-wall nanotubes: Oxygen makes it slip, hydrogen makes it stick},
author = {Fabian L. Thiemann and Christoph Schran and Patrick Rowe and Erich A. Müller and Angelos Michaelides},
doi = {10.1021/acsnano.2c02784},
year = {2022},
date = {2022-06-01},
urldate = {2022-06-01},
journal = {ACS Nano},
volume = {16},
number = {7},
pages = {10775–10782},
abstract = {Experimental measurements have reported ultra-fast and radius-dependent water transport in carbon nanotubes which are absent in boron nitride nanotubes. Despite considerable effort, the origin of this contrasting (and fascinating) behaviour is not understood. Here, with the aid of machine learning-based molecular dynamics simulations that deliver first-principles accuracy, we investigate water transport in single-wall carbon and boron nitride nanotubes. Our simulations reveal a large, radius-dependent hydrodynamic slippage on both materials with water experiencing indeed a $backslashapprox 5$ times lower friction on carbon surfaces compared to boron nitride. Analysis of the diffusion mechanisms across the two materials reveals that the fast water transport on carbon is governed by facile oxygen motion, whereas the higher friction on boron nitride arises from specific hydrogen-nitrogen interactions. This work not only delivers a clear reference of unprecedented accuracy for water flow in single-wall nanotubes, but also provides detailed mechanistic insight into its radius and material dependence for future technological application.},
keywords = {Confinement, Hydrogen bonding, Machine Learning Potentials, Water at Interfaces, Water flow},
pubstate = {published},
tppubtype = {article}
}
2021
Christoph Schran, Fabian L. Thiemann, Patrick Rowe, Erich A. Müller, Ondrej Marsalek, Angelos Michaelides
Machine learning potentials for complex aqueous systems made simple Journal Article
In: Proc. Natl. Acad. Sci., vol. 118, no. 38, pp. e2110077118, 2021, ISSN: 0027-8424.
Abstract | Links | BibTeX | Tags: Confinement, Ions in Water, Machine Learning Potentials, Water, Water at Interfaces
@article{Schran2021/10.1073/PNAS.2110077118,
title = {Machine learning potentials for complex aqueous systems made simple},
author = {Christoph Schran and Fabian L. Thiemann and Patrick Rowe and Erich A. Müller and Ondrej Marsalek and Angelos Michaelides},
doi = {10.1073/PNAS.2110077118},
issn = {0027-8424},
year = {2021},
date = {2021-09-01},
urldate = {2021-09-01},
journal = {Proc. Natl. Acad. Sci.},
volume = {118},
number = {38},
pages = {e2110077118},
publisher = {National Academy of Sciences},
abstract = {Simulation techniques based on accurate and efficient representations of potential energy surfaces are urgently needed for the understanding of complex aqueous systems such as solid-liquid interfaces. Here, we present a machine learning framework that enables the efficient development and validation of models for complex aqueous systems. Instead of trying to deliver a globally-optimal machine learning potential, we propose to develop models applicable to specific thermodynamic state points in a simple and user-friendly process. After an initial ab initio simulation, a machine learning potential is constructed with minimum human effort through a data-driven active learning protocol. Such models can afterwards be applied in exhaustive simulations to provide reliable answers for the scientific question at hand. We showcase this methodology on a diverse set of aqueous systems with increasing degrees of complexity. The systems chosen here comprise water with different ions in solution, water on a titanium dioxide surface, as well as water confined in nanotubes and between molybdenum disulfide sheets. Highlighting the accuracy of our approach compared to the ab initio reference, the resulting models are evaluated in detail with an automated validation protocol that includes structural and dynamical properties and the precision of the force prediction of the models. Finally, we demonstrate the capabilities of our approach for the description of water on the rutile titanium dioxide (110) surface to analyze structure and mobility of water on this surface. Such machine learning models provide a straightforward and uncomplicated but accurate extension of simulation time and length scales for complex systems.},
keywords = {Confinement, Ions in Water, Machine Learning Potentials, Water, Water at Interfaces},
pubstate = {published},
tppubtype = {article}
}
Christoph Schran, Fabien Brieuc, Dominik Marx
Transferability of machine learning potentials: Protonated water neural network potential applied to the protonated water hexamer Journal Article
In: J. Chem. Phys., vol. 154, no. 5, pp. 051101, 2021, ISSN: 10897690.
Abstract | Links | BibTeX | Tags: Coupled Cluster, Hydrogen bonding, Machine Learning Potentials, Water
@article{Schran2021/10.1063/5.0035438,
title = {Transferability of machine learning potentials: Protonated water neural network potential applied to the protonated water hexamer},
author = {Christoph Schran and Fabien Brieuc and Dominik Marx},
doi = {10.1063/5.0035438},
issn = {10897690},
year = {2021},
date = {2021-02-01},
urldate = {2021-02-01},
journal = {J. Chem. Phys.},
volume = {154},
number = {5},
pages = {051101},
abstract = {A previously published neural network potential for the description of protonated water clusters up to the protonated water tetramer, H+(H2O)4, at an essentially converged coupled cluster accuracy [C. Schran, J. Behler, and D. Marx, J. Chem. Theory Comput. 16, 88 (2020)] is applied to the protonated water hexamer, H+(H2O)6 - a system that the neural network has never seen before. Although being in the extrapolation regime, it is shown that the potential not only allows for quantum simulations from ultra-low temperatures ∼1 K up to 300 K but is also able to describe the new system very accurately compared to explicit coupled cluster calculations. This transferability of the model is rationalized by the similarity of the atomic environments encountered for the larger cluster compared to the environments in the training set of the model. Compared to the interpolation regime, the quality of the model is reduced by roughly one order of magnitude, but most of the difference to the coupled cluster reference comes from global shifts of the potential energy surface, while local energy fluctuations are well recovered. These results suggest that the application of neural network potentials in extrapolation regimes can provide useful results and might be more general than usually thought.},
keywords = {Coupled Cluster, Hydrogen bonding, Machine Learning Potentials, Water},
pubstate = {published},
tppubtype = {article}
}
2020
Christoph Schran, Krystof Brezina, Ondrej Marsalek
Committee neural network potentials control generalization errors and enable active learning Journal Article
In: J. Chem. Phys., vol. 153, no. 10, pp. 104105, 2020, ISSN: 10897690.
Abstract | Links | BibTeX | Tags: Machine Learning Potentials, path integral molecular dynamics (PIMD), Water
@article{Schran2020/10.1063/5.0016004,
title = {Committee neural network potentials control generalization errors and enable active learning},
author = {Christoph Schran and Krystof Brezina and Ondrej Marsalek},
doi = {10.1063/5.0016004},
issn = {10897690},
year = {2020},
date = {2020-09-01},
urldate = {2020-09-01},
journal = {J. Chem. Phys.},
volume = {153},
number = {10},
pages = {104105},
abstract = {It is well known in the field of machine learning that committee models improve accuracy, provide generalization error estimates, and enable active learning strategies. In this work, we adapt these concepts to interatomic potentials based on artificial neural networks. Instead of a single model, multiple models that share the same atomic environment descriptors yield an average that outperforms its individual members as well as a measure of the generalization error in the form of the committee disagreement. We not only use this disagreement to identify the most relevant configurations to build up the model's training set in an active learning procedure but also monitor and bias it during simulations to control the generalization error. This facilitates the adaptive development of committee neural network potentials and their training sets while keeping the number of ab initio calculations to a minimum. To illustrate the benefits of this methodology, we apply it to the development of a committee model for water in the condensed phase. Starting from a single reference ab initio simulation, we use active learning to expand into new state points and to describe the quantum nature of the nuclei. The final model, trained on 814 reference calculations, yields excellent results under a range of conditions, from liquid water at ambient and elevated temperatures and pressures to different phases of ice, and the air-water interface - all including nuclear quantum effects. This approach to committee models will enable the systematic development of robust machine learning models for a broad range of systems.},
keywords = {Machine Learning Potentials, path integral molecular dynamics (PIMD), Water},
pubstate = {published},
tppubtype = {article}
}
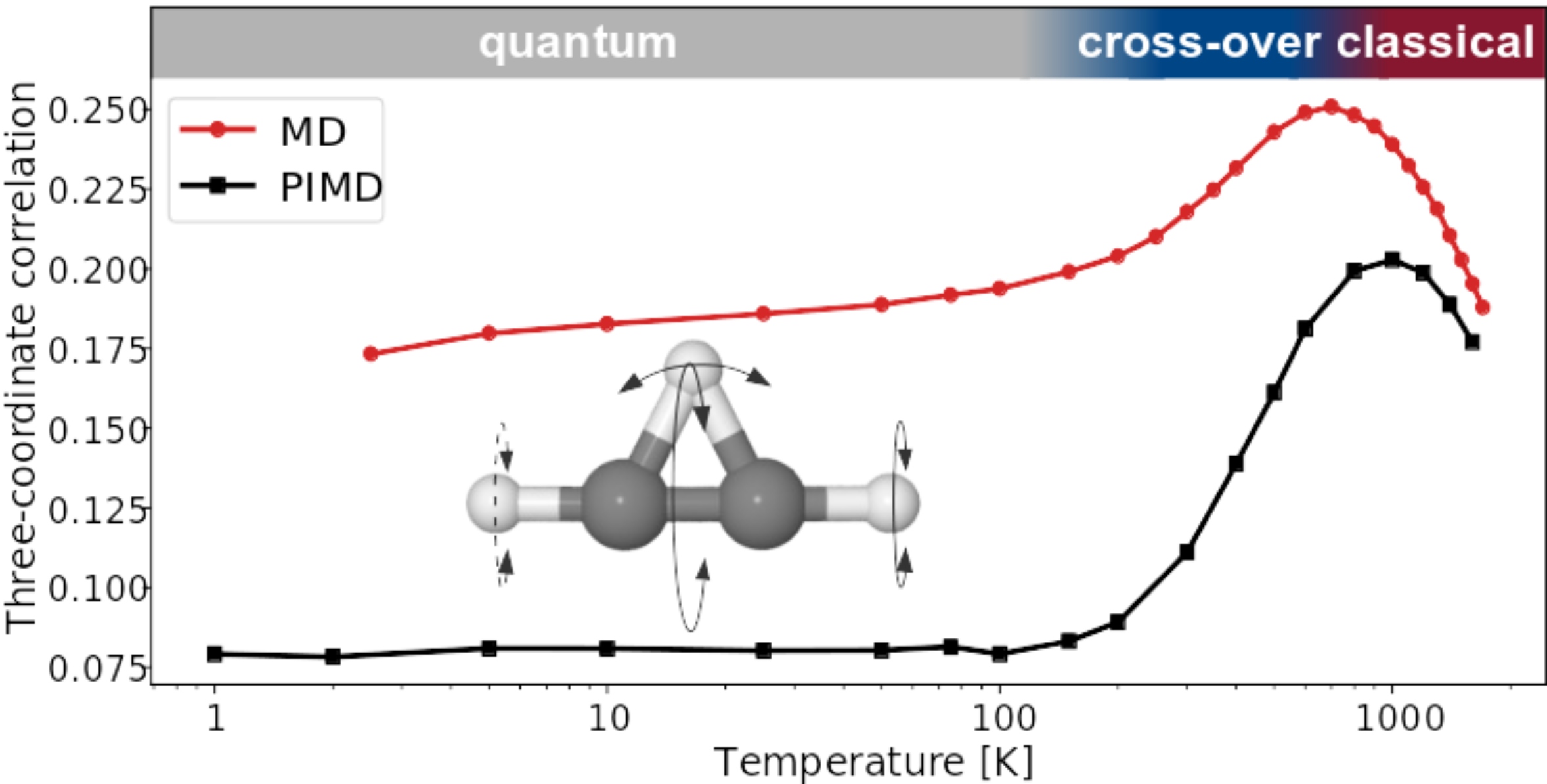
Rafał Topolnicki, Fabien Brieuc, Christoph Schran, Dominik Marx
Deciphering high-order structural correlations within fluxional molecules from classical and quantum configurational entropy Journal Article
In: J. Chem. Theory Comput., vol. 16, no. 11, pp. 6785–6794, 2020, ISSN: 15499626.
Abstract | Links | BibTeX | Tags: Nuclear quantum effects
@article{Topolnicki2020/10.1021/acs.jctc.0c00642,
title = {Deciphering high-order structural correlations within fluxional molecules from classical and quantum configurational entropy},
author = {Rafał Topolnicki and Fabien Brieuc and Christoph Schran and Dominik Marx},
doi = {10.1021/acs.jctc.0c00642},
issn = {15499626},
year = {2020},
date = {2020-09-01},
urldate = {2020-09-01},
journal = {J. Chem. Theory Comput.},
volume = {16},
number = {11},
pages = {6785–6794},
abstract = {We employ the kth nearest-neighbor estimator of configurational entropy in order to decode within a parameter-free numerical approach the complex high-order structural correlations in fluxional molecules going much beyond the usual linear, bivariate correlations. This generic entropy-based scheme for determining many-body correlations is applied to the complex configurational ensemble of protonated acetylene, a prototype for fluxional molecules featuring large-Amplitude motion. After revealing the importance of high-order correlations beyond the simple two-coordinate picture for this molecule, we analyze in detail the evolution of the relevant correlations with temperature as well as the impact of nuclear quantum effects down to the ultralow temperature regime of 1 K. We find that quantum delocalization and zero-point vibrations significantly reduce all correlations in protonated acetylene in the deep quantum regime. Even at low temperatures up to about 100 K, most correlations are essentially absent in the quantum case and only gain importance at higher temperatures. In the high temperature regime, beyond roughly 800 K, the increasing thermal fluctuations are found to exert a destructive effect on the presence of correlations. At intermediate temperatures of approximately 100-800 K, a quantum-To-classical cross-over regime is found where classical mechanics starts to correctly describe trends in the correlations whereas it even qualitatively fails below 100 K. Finally, a classical description of the nuclei provides correlations that are in quantitative agreement with the quantum ones only at temperatures exceeding 1000 K. This data-intensive analysis has been made possible due to recent developments of machine learning techniques based on high-dimensional neural network potential energy surfaces in full dimensionality that allow us to exhaustively sample both the classical and quantum ensemble of protonated acetylene at essentially converged coupled cluster accuracy from 1 to more than 1000 K. The presented non-parametric analysis of correlations beyond usual linear two-coordinate terms is transferable to other system classes. The technique is also expected to complement and guide the analysis of experimental measurements, in particular multidimensional vibrational spectroscopy, by revealing the complex coupling between various degrees of freedom.},
keywords = {Nuclear quantum effects},
pubstate = {published},
tppubtype = {article}
}
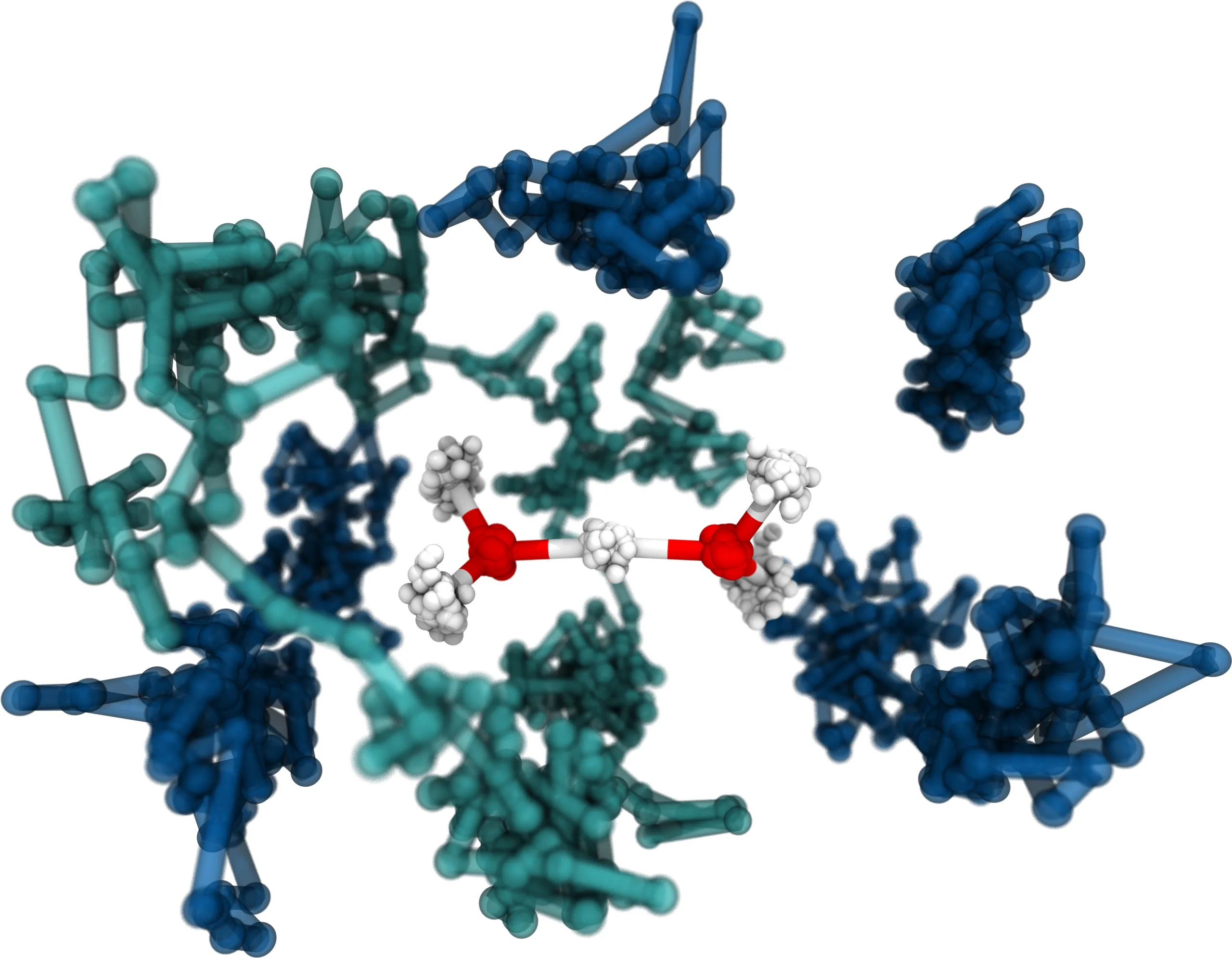
Fabien Brieuc, Christoph Schran, Felix Uhl, Harald Forbert, Dominik Marx
Converged quantum simulations of reactive solutes in superfluid helium: The Bochum perspective Journal Article
In: J. Chem. Phys., vol. 152, no. 21, pp. 210901, 2020, ISSN: 10897690.
Abstract | Links | BibTeX | Tags: Nuclear quantum effects, path integral molecular dynamics (PIMD), Superfluidity
@article{Brieuc2020/10.1063/5.0008309,
title = {Converged quantum simulations of reactive solutes in superfluid helium: The Bochum perspective},
author = {Fabien Brieuc and Christoph Schran and Felix Uhl and Harald Forbert and Dominik Marx},
doi = {10.1063/5.0008309},
issn = {10897690},
year = {2020},
date = {2020-06-01},
urldate = {2020-06-01},
journal = {J. Chem. Phys.},
volume = {152},
number = {21},
pages = {210901},
abstract = {Superfluid helium has not only fascinated scientists for centuries but is also the ideal matrix for the investigation of chemical systems under ultra-cold conditions in helium nanodroplet isolation experiments. Together with related experimental techniques such as helium tagging photodissociation spectroscopy, these methods have provided unique insights into many interesting systems. Complemented by theoretical work, they were additionally able to greatly expand our general understanding of manifestations of superfluid behavior in finite sized clusters and their response to molecular impurities. However, most theoretical studies up to now have not included the reactivity and flexibility of molecular systems embedded in helium. In this perspective, the theoretical foundation of simulating fluxional molecules and reactive complexes in superfluid helium is presented in detail. Special emphasis is put on recent developments for the converged description of both the molecular interactions and the quantum nature of the nuclei at ultra-low temperatures. As a first step, our hybrid path integral molecular dynamics/bosonic path integral Monte Carlo method is reviewed. Subsequently, methods for efficient path integral sampling tailored for this hybrid coupling scheme are discussed while also introducing new developments to enhance the accurate incorporation of the solute⋯solvent coupling. Finally, highly accurate descriptions of the interactions in solute⋯helium systems using machine learning techniques are addressed. Our current automated and adaptive fitting procedures to parameterize high-dimensional neural network potentials for both the full-dimensional potential energy surface of solutes and the solute⋯solvent interaction potentials are concisely presented. They are demonstrated to faithfully represent many-body potential functions able to describe chemically complex and reactive solutes in helium environments seamlessly from one He atom up to bulk helium at the accuracy level of coupled cluster electronic structure calculations. Together, these advances allow for converged quantum simulations of fluxional and reactive solutes in superfluid helium under cryogenic conditions.},
keywords = {Nuclear quantum effects, path integral molecular dynamics (PIMD), Superfluidity},
pubstate = {published},
tppubtype = {article}
}
Christoph Schran, Jörg Behler, Dominik Marx
Automated fitting of neural network potentials at coupled cluster accuracy: Protonated water clusters as testing ground Journal Article
In: J. Chem. Theory Comput., vol. 16, no. 1, pp. 88–99, 2020, ISSN: 15499626.
Abstract | Links | BibTeX | Tags: Coupled Cluster, Hydrogen bonding, Machine Learning Potentials, Water
@article{Schran2020/10.1021/acs.jctc.9b00805,
title = {Automated fitting of neural network potentials at coupled cluster accuracy: Protonated water clusters as testing ground},
author = {Christoph Schran and Jörg Behler and Dominik Marx},
doi = {10.1021/acs.jctc.9b00805},
issn = {15499626},
year = {2020},
date = {2020-01-01},
urldate = {2020-01-01},
journal = {J. Chem. Theory Comput.},
volume = {16},
number = {1},
pages = {88–99},
abstract = {Highly accurate potential energy surfaces are of key interest for the detailed understanding and predictive modeling of chemical systems. In recent years, several new types of force fields, which are based on machine learning algorithms and fitted to ab initio reference calculations, have been introduced to meet this requirement. Here, we show how high-dimensional neural network potentials can be employed to automatically generate the potential energy surface of finite sized clusters at coupled cluster accuracy, namely CCSD(T*)-F12a/aug-cc-pVTZ. The developed automated procedure utilizes the established intrinsic properties of the model such that the configurations for the training set are selected in an unbiased and efficient way to minimize the computational effort of expensive reference calculations. These ideas are applied to protonated water clusters from the hydronium cation, H3O+, up to the tetramer, H9O4+, and lead to a single potential energy surface that describes all these systems at essentially converged coupled cluster accuracy with a fitting error of 0.06 kJ/mol per atom. The fit is validated in detail for all clusters up to the tetramer and yields reliable results not only for stationary points but also for reaction pathways and intermediate configurations as well as different sampling techniques. Per design, the neural network potentials (NNPs) constructed in this fashion can handle very different conditions including the quantum nature of the nuclei and enhanced sampling techniques covering very low as well as high temperatures. This enables fast and exhaustive exploration of the targeted protonated water clusters with essentially converged interactions. In addition, the automated process will allow one to tackle finite systems much beyond the present case.},
keywords = {Coupled Cluster, Hydrogen bonding, Machine Learning Potentials, Water},
pubstate = {published},
tppubtype = {article}
}
2019
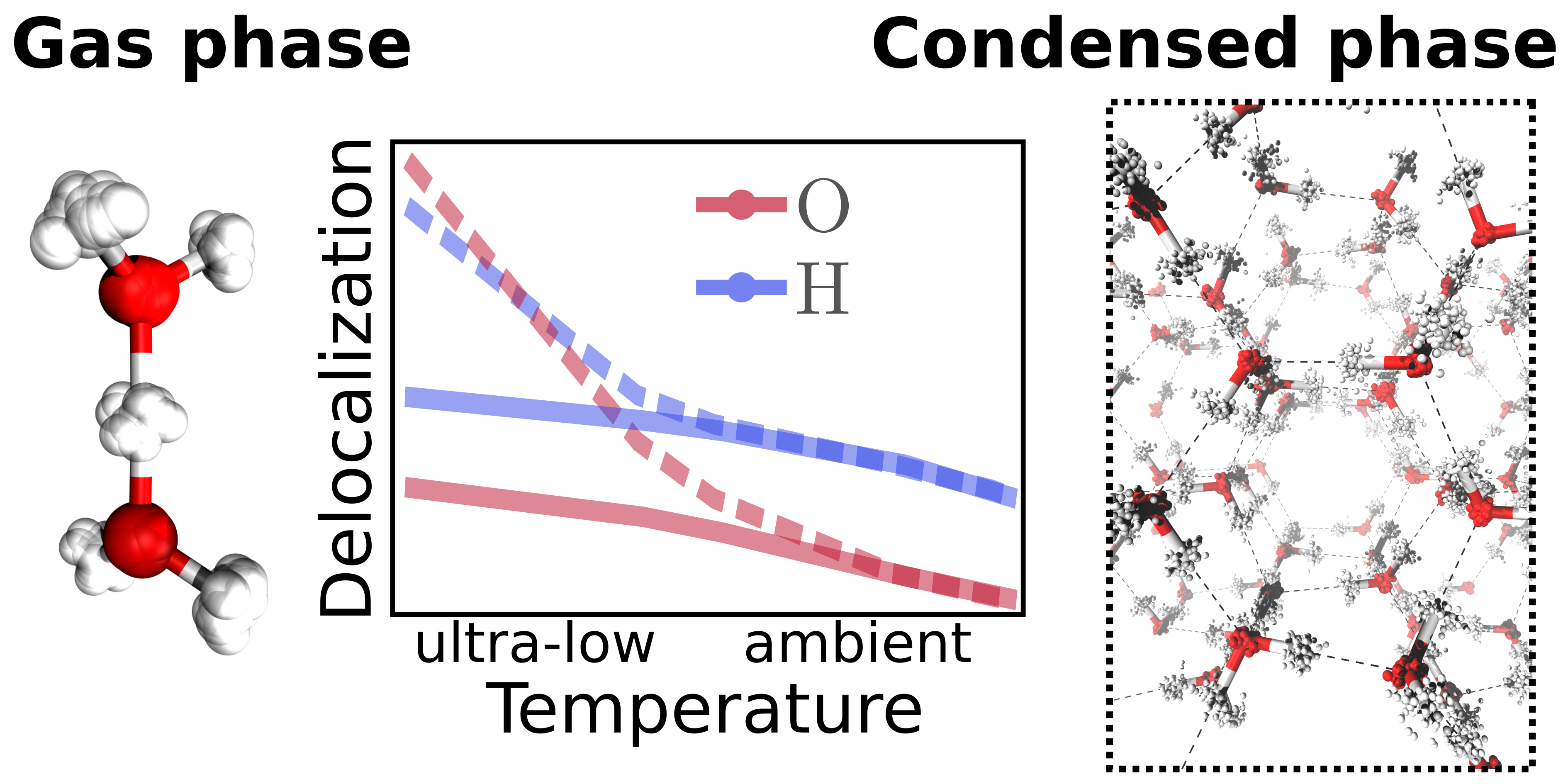
Christoph Schran, Dominik Marx
Quantum nature of the hydrogen bond from ambient conditions down to ultra-low temperatures Journal Article
In: Phys. Chem. Chem. Phys., vol. 21, no. 45, pp. 24967–24975, 2019, ISSN: 14639076.
Abstract | Links | BibTeX | Tags: Hydrogen bonding, Nuclear quantum effects, path integral molecular dynamics (PIMD), Water
@article{Schran2019/10.1039/C9CP04795F,
title = {Quantum nature of the hydrogen bond from ambient conditions down to ultra-low temperatures},
author = {Christoph Schran and Dominik Marx},
doi = {10.1039/c9cp04795f},
issn = {14639076},
year = {2019},
date = {2019-10-01},
urldate = {2019-10-01},
journal = {Phys. Chem. Chem. Phys.},
volume = {21},
number = {45},
pages = {24967–24975},
abstract = {Many experimental techniques such as tagging photodissociation and helium nanodroplet isolation spectroscopy operate at very low temperatures in order to investigate hydrogen bonding. To elucidate the differences between such ultra-cold and usual ambient conditions, different hydrogen bonded systems are studied systematically from 300 K down to about 1 K using path integral simulations that explicitly consider both the quantum nature of the nuclei and thermal fluctuations. For this purpose, finite sized water clusters, specifically the water dimer and hexamer, protonated water clusters including the Zundel and Eigen complexes, as well as hexagonal ice as a condensed phase representative are compared directly as a function of temperature. While weaker hydrogen bonds, as present in the neutral systems, show distinct structural differences between ambient conditions and the ultra-cold regime, the stronger hydrogen bonds of the protonated water clusters are less perturbed by temperature compared to their quantum ground state. In all the studied systems, the quantum delocalization of the nuclei is found to vary drastically with temperature. Interestingly, upon reaching temperatures of about 1 K, the spatial quantum delocalization of the heavy oxygens approaches that of the protons for relatively weak spatial constraints, and even significantly exceeds the latter in the case of the centered hydrogen bond in the Zundel complex. These findings are relevant for comparisons between experiments on hydrogen bonding carried out under ultra-cold versus ambient conditions as well as to understand quantum delocalization phenomena of nuclei by seamlessly extending our insights into noncovalent interactions down to ultra-low temperatures.},
keywords = {Hydrogen bonding, Nuclear quantum effects, path integral molecular dynamics (PIMD), Water},
pubstate = {published},
tppubtype = {article}
}
2018
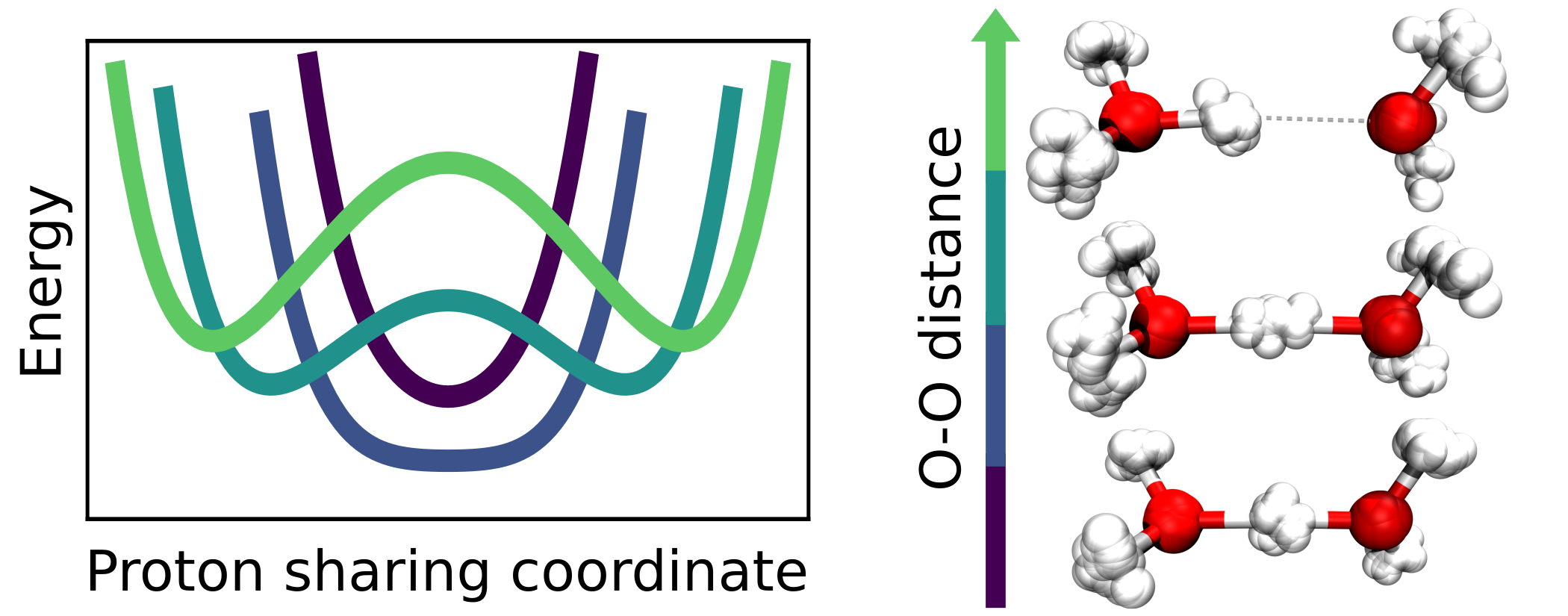
Christoph Schran, Fabien Brieuc, Dominik Marx
Converged colored noise path integral molecular dynamics study of the zundel cation down to ultralow temperatures at coupled cluster accuracy Journal Article
In: J. Chem. Theory Comput., vol. 14, no. 10, pp. 5068–5078, 2018, ISSN: 15499626.
Abstract | Links | BibTeX | Tags: Nuclear quantum effects, path integral molecular dynamics (PIMD), Water
@article{Schran2018/10.1021/acs.jctc.8b00705,
title = {Converged colored noise path integral molecular dynamics study of the zundel cation down to ultralow temperatures at coupled cluster accuracy},
author = {Christoph Schran and Fabien Brieuc and Dominik Marx},
doi = {10.1021/acs.jctc.8b00705},
issn = {15499626},
year = {2018},
date = {2018-09-01},
urldate = {2018-09-01},
journal = {J. Chem. Theory Comput.},
volume = {14},
number = {10},
pages = {5068–5078},
abstract = {For a long time, performing converged path integral simulations at ultra-low, but finite temperatures of a few Kelvin has been a nearly impossible task. However, recent developments in advanced colored noise thermostatting schemes for path integral simulations, namely the Path Integral Generalized Langevin Equation Thermostat (PIGLET) and the Path Integral Quantum Thermal Bath (PIQTB), have been able to greatly reduce the computational cost of these simulations, thus making the ultra-low temperature regime accessible in practice. In this work, we investigate the influence of these two thermostatting schemes on the description of hydrogen-bonded systems at temperatures down to a few Kelvin as encountered, for example, in helium nanodroplet isolation or tagging photodissociation spectroscopy experiments. For this purpose, we analyze the prototypical hydrogen bond in the Zundel cation (H5O2+) as a function of both, oxygen-oxygen distance and temperature in order to elucidate how the anisotropic quantum deloc...},
keywords = {Nuclear quantum effects, path integral molecular dynamics (PIMD), Water},
pubstate = {published},
tppubtype = {article}
}
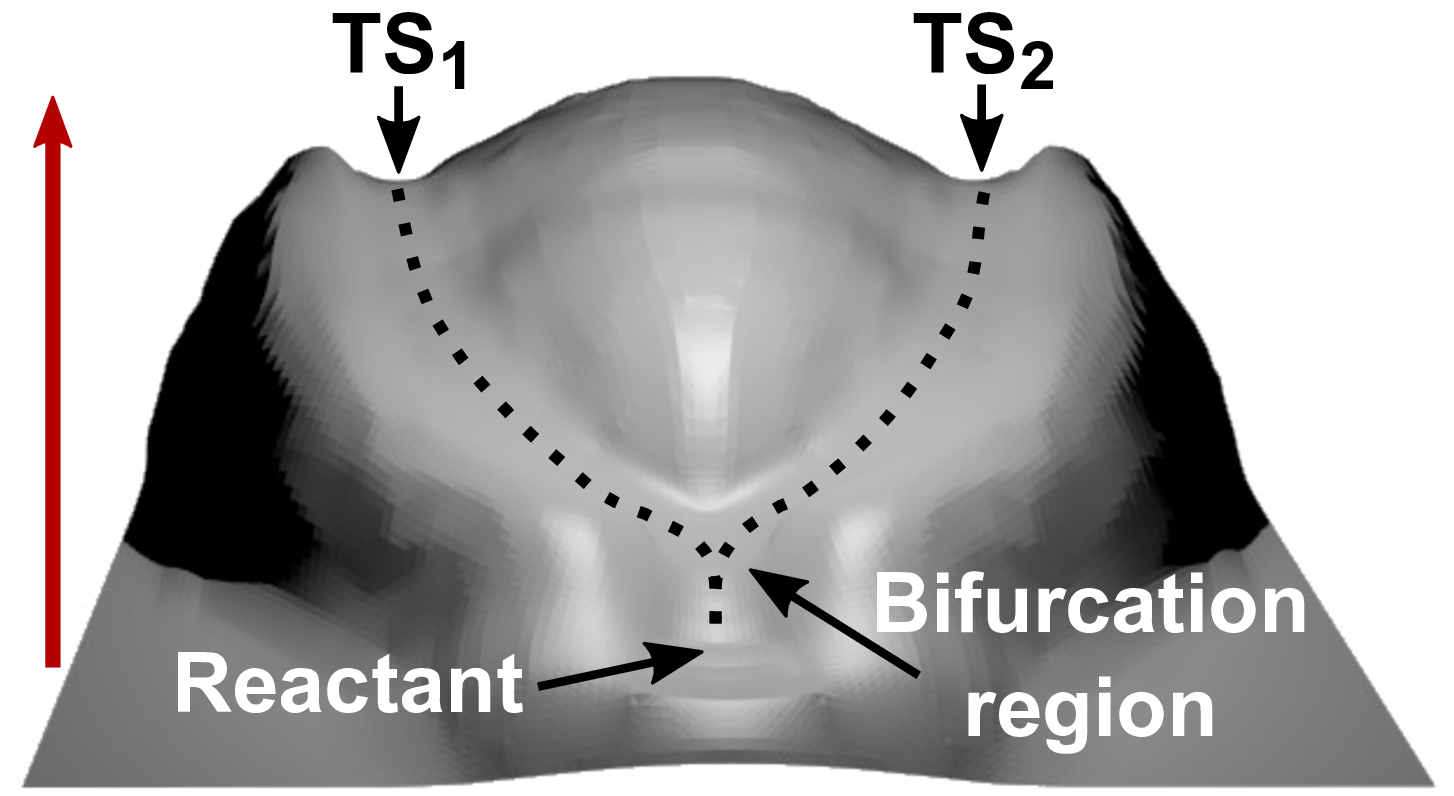
Miriam Wollenhaupt, Christoph Schran, Martin Krupička, Dominik Marx
In: ChemPhysChem, vol. 19, no. 7, pp. 837–847, 2018, ISSN: 14397641.
Abstract | Links | BibTeX | Tags: AIMD, Potential Energy Surface
@article{Wollenhaupt2018/10.1002/cphc.201701209,
title = {Force-induced catastrophes on energy landscapes: Mechanochemical manipulation of downhill and uphill bifurcations explains the ring-opening selectivity of cyclopropanes},
author = {Miriam Wollenhaupt and Christoph Schran and Martin Krupička and Dominik Marx},
doi = {10.1002/cphc.201701209},
issn = {14397641},
year = {2018},
date = {2018-04-01},
urldate = {2018-04-01},
journal = {ChemPhysChem},
volume = {19},
number = {7},
pages = {837–847},
abstract = {The mechanochemistry of ring-opening reactions of cyclopropane derivatives turns out to be unexpectedly rich and puzzling. After showing that a rare so-called uphill bifurcation in the case of trans-gem-difluorocyclopropane turns into a downhill bifurcation upon substitution of fluorine by chlorine, bromine, and iodine in the thermal activation limit, the dichloro derivative is studied systematically in the realm of mechanochemical activation. Detailed exploration of the force-transformed potential energy surface of trans-gem-dichlorocyclopropane in terms of Dijkstra path analysis unveils a hitherto unknown topological catastrophe where the global shape of the energy landscape is fundamentally changed. From thermal activation up to moderately large forces, it is an uphill bifurcation that decides about dis- versus conrotatory ring-opening followed by separate transition states along both pathways. Above a critical force, the two distinct transition states merge to yield a single transition state such that the decision about the dis- versus conrotatory ring-opening process is taken at a newly established downhill bifurcation. The discovery of a force-induced qualitative change of the topology of a reaction network vastly transcends the previous understanding of the ring-opening reaction of this species. It would be astonishing to not discover a wealth of such catastrophes for mechanochemically activated reactions, which will greatly extend the known opportunities to manipulate chemical reaction networks.},
keywords = {AIMD, Potential Energy Surface},
pubstate = {published},
tppubtype = {article}
}
Christoph Schran, Felix Uhl, Jörg Behler, Dominik Marx
High-dimensional neural network potentials for solvation: The case of protonated water clusters in helium Journal Article
In: J. Chem. Phys., vol. 148, no. 10, pp. 102310, 2018, ISSN: 00219606.
Abstract | Links | BibTeX | Tags: Machine Learning Potentials, Nuclear quantum effects, Superfluidity, Water
@article{Schran2018/10.1063/1.4996819,
title = {High-dimensional neural network potentials for solvation: The case of protonated water clusters in helium},
author = {Christoph Schran and Felix Uhl and Jörg Behler and Dominik Marx},
doi = {10.1063/1.4996819},
issn = {00219606},
year = {2018},
date = {2018-03-01},
urldate = {2018-03-01},
journal = {J. Chem. Phys.},
volume = {148},
number = {10},
pages = {102310},
abstract = {The design of accurate helium-solute interaction potentials for the simulation of chemically complex molecules solvated in superfluid helium has long been a cumbersome task due to the rather weak but strongly anisotropic nature of the interactions. We show that this challenge can be met by using a combination of an effective pair potential for the He–He interactions and a flexible high-dimensional neural network potential (NNP) for describing the complex interaction between helium and the solute in a pairwise additive manner. This approach yields an excellent agreement with a mean absolute deviation as small as 0.04 kJ mol−1 for the interaction energy between helium and both hydronium and Zundel cations compared with coupled cluster reference calculations with an energetically converged basis set. The construction and improvement of the potential can be performed in a highly automated way, which opens the door for applications to a variety of reactive molecules to study the effect of solvation on the solu...},
keywords = {Machine Learning Potentials, Nuclear quantum effects, Superfluidity, Water},
pubstate = {published},
tppubtype = {article}
}
2017
Christoph Schran, Ondrej Marsalek, Thomas E. Markland
Unravelling the influence of quantum proton delocalization on electronic charge transfer through the hydrogen bond Journal Article
In: Chem. Phys. Lett., vol. 678, pp. 289–295, 2017, ISSN: 00092614.
Abstract | Links | BibTeX | Tags: Charge transfer, Hydrogen bonding, Ions in Water, Nuclear quantum effects, Water
@article{Schran2017/10.1016/j.cplett.2017.04.034,
title = {Unravelling the influence of quantum proton delocalization on electronic charge transfer through the hydrogen bond},
author = {Christoph Schran and Ondrej Marsalek and Thomas E. Markland},
doi = {10.1016/j.cplett.2017.04.034},
issn = {00092614},
year = {2017},
date = {2017-06-01},
urldate = {2017-06-01},
journal = {Chem. Phys. Lett.},
volume = {678},
pages = {289–295},
abstract = {Upon hydrogen bond formation, electronic charge density is transferred between the donor and acceptor, impacting processes ranging from hydration to spectroscopy. Here we use ab initio path integral simulations to elucidate the role of nuclear quantum effects in determining the charge transfer in a range of hydrogen bonded species in the gas and liquid phase. We show that the quantization of the nuclei gives rise to large changes in the magnitude of the charge transfer as well as its temperature dependence. We then explain how a single geometric parameter determines the charge transfer through the hydrogen bond. These results thus demonstrate that nuclear quantum effects are vital for the accurate description of charge transfer and offer a physically transparent way to understand how hydrogen bonding gives rise to it.},
keywords = {Charge transfer, Hydrogen bonding, Ions in Water, Nuclear quantum effects, Water},
pubstate = {published},
tppubtype = {article}
}
2016
Maciej Śmiechowski, Christoph Schran, Harald Forbert, Dominik Marx
Correlated particle motion and THz spectral response of supercritical water Journal Article
In: Phys. Rev. Lett., vol. 116, no. 2, 2016, ISSN: 10797114.
Abstract | Links | BibTeX | Tags: Hydrogen bonding, Spectra, Water
@article{Smiechowski2016/10.1103/PhysRevLett.116.027801,
title = {Correlated particle motion and THz spectral response of supercritical water},
author = {Maciej Śmiechowski and Christoph Schran and Harald Forbert and Dominik Marx},
doi = {10.1103/PhysRevLett.116.027801},
issn = {10797114},
year = {2016},
date = {2016-01-01},
urldate = {2016-01-01},
journal = {Phys. Rev. Lett.},
volume = {116},
number = {2},
abstract = {Molecular dynamics simulations of supercritical water reveal distinctly different distance-dependent modulations of dipolar response and correlations in particle motion compared to ambient conditions. The strongly perturbed H-bond network of water at supercritical conditions allows for considerable translational and rotational freedom of individual molecules. These changes give rise to substantially different infrared spectra and vibrational density of states at THz frequencies for densities above and below the Widom line that separates percolating liquidlike and clustered gaslike supercritical water.},
keywords = {Hydrogen bonding, Spectra, Water},
pubstate = {published},
tppubtype = {article}
}