2025
Hubert Beck, Pavol Simko, Lars L. Schaaf, Ondrej Marsalek, Christoph Schran
Multi-head committees enable direct uncertainty prediction for atomistic foundation models Journal Article
In: J. Chem. Phys., vol. 163, iss. 23, pp. 234103, 2025.
Abstract | Links | BibTeX | Tags: Machine Learning Potentials, Potential Energy Surface
@article{Beck2025/10.1063/5.0302097,
title = {Multi-head committees enable direct uncertainty prediction for atomistic foundation models},
author = {Hubert Beck and Pavol Simko and Lars L. Schaaf and Ondrej Marsalek and Christoph Schran},
url = {https://pubs.aip.org/aip/jcp/article/163/23/234103/3374754},
doi = {10.1063/5.0302097},
year = {2025},
date = {2025-12-15},
urldate = {2025-12-15},
journal = {J. Chem. Phys.},
volume = {163},
issue = {23},
pages = {234103},
abstract = {Machine learning potentials have become a standard tool for atomistic materials modeling. While models continue to become more generalizable, an open challenge relates to efficient uncertainty predictions for active learning and robust error analysis. In this work, we utilize MACE and its multi-head mechanism to implement a committee neural network potential for message-passing architectures, where the committee comprises multiple output modules attached to the same atomic environment descriptors. As with traditional committees of independent networks, the standard deviation of the predictions functions as an estimate of the model's uncertainty. We show for a range of datasets in custom-build models that the uncertainty of the force predictions correlates well with the true errors. We subsequently apply this concept to foundation models, in particular MACE-MP-0, where we train only the newly attached output heads while keeping the remaining part of the model fixed. We use this approach in an active learning workflow to condense the training set of the foundation model to just 5% of its original size. The foundation model multi-head committee trained on the condensed training set enables reliable uncertainty estimation without any substantial decrease in prediction accuracy.},
keywords = {Machine Learning Potentials, Potential Energy Surface},
pubstate = {published},
tppubtype = {article}
}
2024

Fabian L. Thiemann, Niamh O’Neill, Venkat Kapil, Angelos Michaelides, Christoph Schran
Introduction to machine learning potentials for atomistic simulations Journal Article
In: J. Phys.: Condens. Matter, vol. 37, no. 7, pp. 073002, 2024.
Abstract | Links | BibTeX | Tags: Machine Learning Potentials, Potential Energy Surface
@article{Thiemann2024/10.1088/1361-648X/ad9657,
title = {Introduction to machine learning potentials for atomistic simulations},
author = {Fabian L. Thiemann and Niamh O’Neill and Venkat Kapil and Angelos Michaelides and Christoph Schran},
url = {https://iopscience.iop.org/article/10.1088/1361-648X/ad9657/meta},
doi = {10.1088/1361-648X/ad9657},
year = {2024},
date = {2024-12-06},
journal = {J. Phys.: Condens. Matter},
volume = {37},
number = {7},
pages = {073002},
abstract = {Machine learning potentials have revolutionised the field of atomistic simulations in recent years and are becoming a mainstay in the toolbox of computational scientists. This paper aims to provide an overview and introduction into machine learning potentials and their practical application to scientific problems. We provide a systematic guide for developing machine learning potentials, reviewing chemical descriptors, regression models, data generation and validation approaches. We begin with an emphasis on the earlier generation of models, such as high-dimensional neural network potentials and Gaussian approximation potentials, to provide historical perspective and guide the reader towards the understanding of recent developments, which are discussed in detail thereafter. Furthermore, we refer to relevant expert reviews, open-source software, and practical examples—further lowering the barrier to exploring these methods. The paper ends with selected showcase examples, highlighting the capabilities of machine learning potentials and how they can be applied to push the boundaries in atomistic simulations.},
keywords = {Machine Learning Potentials, Potential Energy Surface},
pubstate = {published},
tppubtype = {article}
}
2018
Miriam Wollenhaupt, Christoph Schran, Martin Krupička, Dominik Marx
In: ChemPhysChem, vol. 19, no. 7, pp. 837–847, 2018, ISSN: 14397641.
Abstract | Links | BibTeX | Tags: Ab-Initio Molecular Dynamics (AIMD), Potential Energy Surface
@article{Wollenhaupt2018/10.1002/cphc.201701209,
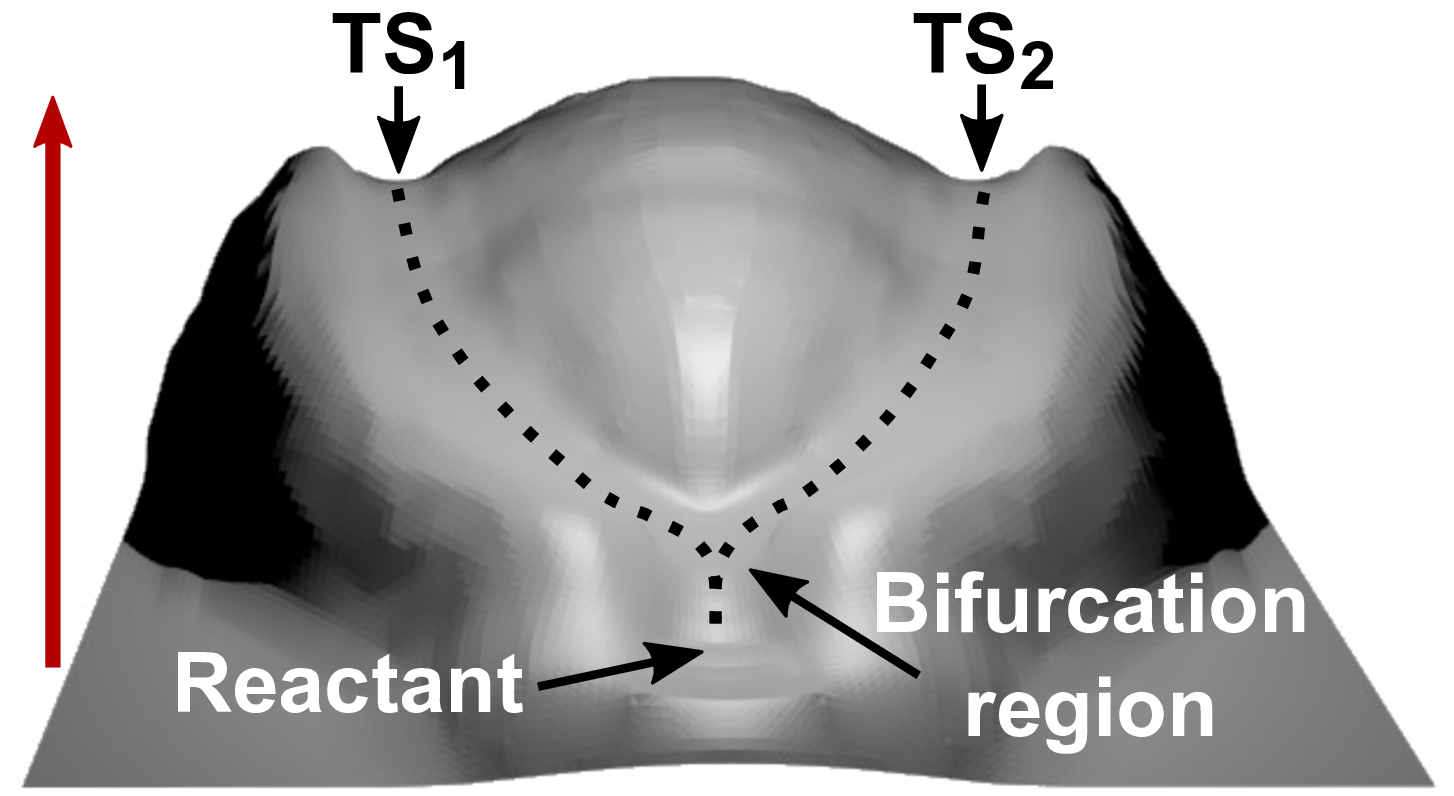
title = {Force-induced catastrophes on energy landscapes: Mechanochemical manipulation of downhill and uphill bifurcations explains the ring-opening selectivity of cyclopropanes},
author = {Miriam Wollenhaupt and Christoph Schran and Martin Krupička and Dominik Marx},
doi = {10.1002/cphc.201701209},
issn = {14397641},
year = {2018},
date = {2018-04-01},
urldate = {2018-04-01},
journal = {ChemPhysChem},
volume = {19},
number = {7},
pages = {837–847},
abstract = {The mechanochemistry of ring-opening reactions of cyclopropane derivatives turns out to be unexpectedly rich and puzzling. After showing that a rare so-called uphill bifurcation in the case of trans-gem-difluorocyclopropane turns into a downhill bifurcation upon substitution of fluorine by chlorine, bromine, and iodine in the thermal activation limit, the dichloro derivative is studied systematically in the realm of mechanochemical activation. Detailed exploration of the force-transformed potential energy surface of trans-gem-dichlorocyclopropane in terms of Dijkstra path analysis unveils a hitherto unknown topological catastrophe where the global shape of the energy landscape is fundamentally changed. From thermal activation up to moderately large forces, it is an uphill bifurcation that decides about dis- versus conrotatory ring-opening followed by separate transition states along both pathways. Above a critical force, the two distinct transition states merge to yield a single transition state such that the decision about the dis- versus conrotatory ring-opening process is taken at a newly established downhill bifurcation. The discovery of a force-induced qualitative change of the topology of a reaction network vastly transcends the previous understanding of the ring-opening reaction of this species. It would be astonishing to not discover a wealth of such catastrophes for mechanochemically activated reactions, which will greatly extend the known opportunities to manipulate chemical reaction networks.},
keywords = {Ab-Initio Molecular Dynamics (AIMD), Potential Energy Surface},
pubstate = {published},
tppubtype = {article}
}