2026

Marcella Iannuzzi, Jan Wilhelm, Frederick Stein, Augustin Bussy, Hossam Elgabarty, Dorothea Golze, Anna-Sophia Hehn, Maximilian Graml, Stepan Marek, Beliz Sertcan Gökmen, Christoph Schran, Harald Forbert, Rustam Z. Khaliullin, Anton Kozhevnikov, Mathieu Taillefumier, Rocco Meli, Vladimir V. Rybkin, Martin Brehm, Robert Schade, Ole Schütt, Johann V. Pototschnig, Hossein Mirhosseini, Andreas Knüpfer, Dominik Marx, Matthias Krack, Jürg Hutter, Thomas D. Kühne
The CP2K program package made simple Journal Article
In: J. Phys. Chem. B, vol. 130, iss. 4, pp. 1237–1310, 2026.
Abstract | Links | BibTeX | Tags: Ab-Initio Molecular Dynamics (AIMD), Density Functional Theory (DFT), Quantum Dynamics
@article{Iannuzzi2026/10.1021/acs.jpcb.5c05851,
title = {The CP2K program package made simple},
author = {Marcella Iannuzzi and Jan Wilhelm and Frederick Stein and Augustin Bussy and Hossam Elgabarty and Dorothea Golze and Anna-Sophia Hehn and Maximilian Graml and Stepan Marek and Beliz Sertcan Gökmen and Christoph Schran and Harald Forbert and Rustam Z. Khaliullin and Anton Kozhevnikov and Mathieu Taillefumier and Rocco Meli and Vladimir V. Rybkin and Martin Brehm and Robert Schade and Ole Schütt and Johann V. Pototschnig and Hossein Mirhosseini and Andreas Knüpfer and Dominik Marx and Matthias Krack and Jürg Hutter and Thomas D. Kühne},
url = {https://pubs.acs.org/doi/full/10.1021/acs.jpcb.5c05851},
doi = {10.1021/acs.jpcb.5c05851},
year = {2026},
date = {2026-01-15},
urldate = {2026-01-15},
journal = {J. Phys. Chem. B},
volume = {130},
issue = {4},
pages = {1237–1310},
abstract = {CP2K is a versatile open-source software package for simulations across a wide range of atomistic systems, from isolated molecules in the gas phase to low-dimensional functional materials and interfaces, as well as highly symmetric crystalline solids, disordered amorphous glasses, and weakly interacting soft-matter systems in the liquid state and in solution. This review highlights CP2K's capabilities for computing both static and dynamical properties using quantum-mechanical and classical simulation methods. In contrast to the accompanying theory and code paper [J. Chem. Phys. 152, 194103 (2020)], the focus here is on the practical usage and applications of CP2K, with underlying theoretical concepts introduced only as needed.},
keywords = {Ab-Initio Molecular Dynamics (AIMD), Density Functional Theory (DFT), Quantum Dynamics},
pubstate = {published},
tppubtype = {article}
}
2024
Thomas Dufils, Christoph Schran, Ji Chen, Andre K. Geim, Laura Fumagalli, Angelos Michaelides
Origin of dielectric polarization suppression in confined water from first principles Journal Article
In: Chem. Sci., vol. 15, iss. 2, pp. 516–527, 2024.
Abstract | Links | BibTeX | Tags: Ab-Initio Molecular Dynamics (AIMD), Confinement, Hydrogen Bonding, Water
@article{Dufils2024/10.1039/D3SC04740G,
title = {Origin of dielectric polarization suppression in confined water from first principles},
author = {Thomas Dufils and Christoph Schran and Ji Chen and Andre K. Geim and Laura Fumagalli and Angelos Michaelides},
url = {http://dx.doi.org/10.1039/D3SC04740G},
doi = {10.1039/D3SC04740G},
year = {2024},
date = {2024-01-01},
urldate = {2024-01-01},
journal = {Chem. Sci.},
volume = {15},
issue = {2},
pages = {516–527},
publisher = {The Royal Society of Chemistry},
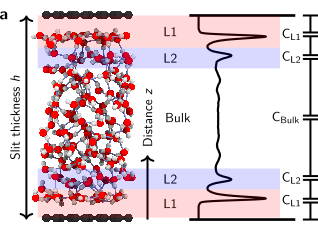
abstract = {It has long been known that the dielectric constant of confined water should be different from that in bulk. Recent experiments have shown that it is vanishingly small, however the origin of the phenomenon remains unclear. Here we used ab initio molecular dynamics simulations (AIMD) and AIMD-trained machine-learning potentials to understand water's structure and electronic properties underpinning this effect. For the graphene and hexagonal boron-nitride substrates considered, we find that it originates in the spontaneous anti-parallel alignment of the water dipoles in the first two water layers near the solid interface. The interfacial layers exhibit net ferroelectric ordering, resulting in an overall anti-ferroelectric arrangement of confined water. Together with constrained hydrogen-bonding orientations, this leads to much reduced out-of-plane polarization. Furthermore, we directly contrast AIMD and simple classical force-field simulations, revealing important differences. This work offers insight into a property of water that is critical in modulating surface forces, the electric-double-layer formation and molecular solvation, and shows a way to compute it.},
keywords = {Ab-Initio Molecular Dynamics (AIMD), Confinement, Hydrogen Bonding, Water},
pubstate = {published},
tppubtype = {article}
}
2018
Miriam Wollenhaupt, Christoph Schran, Martin Krupička, Dominik Marx
In: ChemPhysChem, vol. 19, no. 7, pp. 837–847, 2018, ISSN: 14397641.
Abstract | Links | BibTeX | Tags: Ab-Initio Molecular Dynamics (AIMD), Potential Energy Surface
@article{Wollenhaupt2018/10.1002/cphc.201701209,
title = {Force-induced catastrophes on energy landscapes: Mechanochemical manipulation of downhill and uphill bifurcations explains the ring-opening selectivity of cyclopropanes},
author = {Miriam Wollenhaupt and Christoph Schran and Martin Krupička and Dominik Marx},
doi = {10.1002/cphc.201701209},
issn = {14397641},
year = {2018},
date = {2018-04-01},
urldate = {2018-04-01},
journal = {ChemPhysChem},
volume = {19},
number = {7},
pages = {837–847},
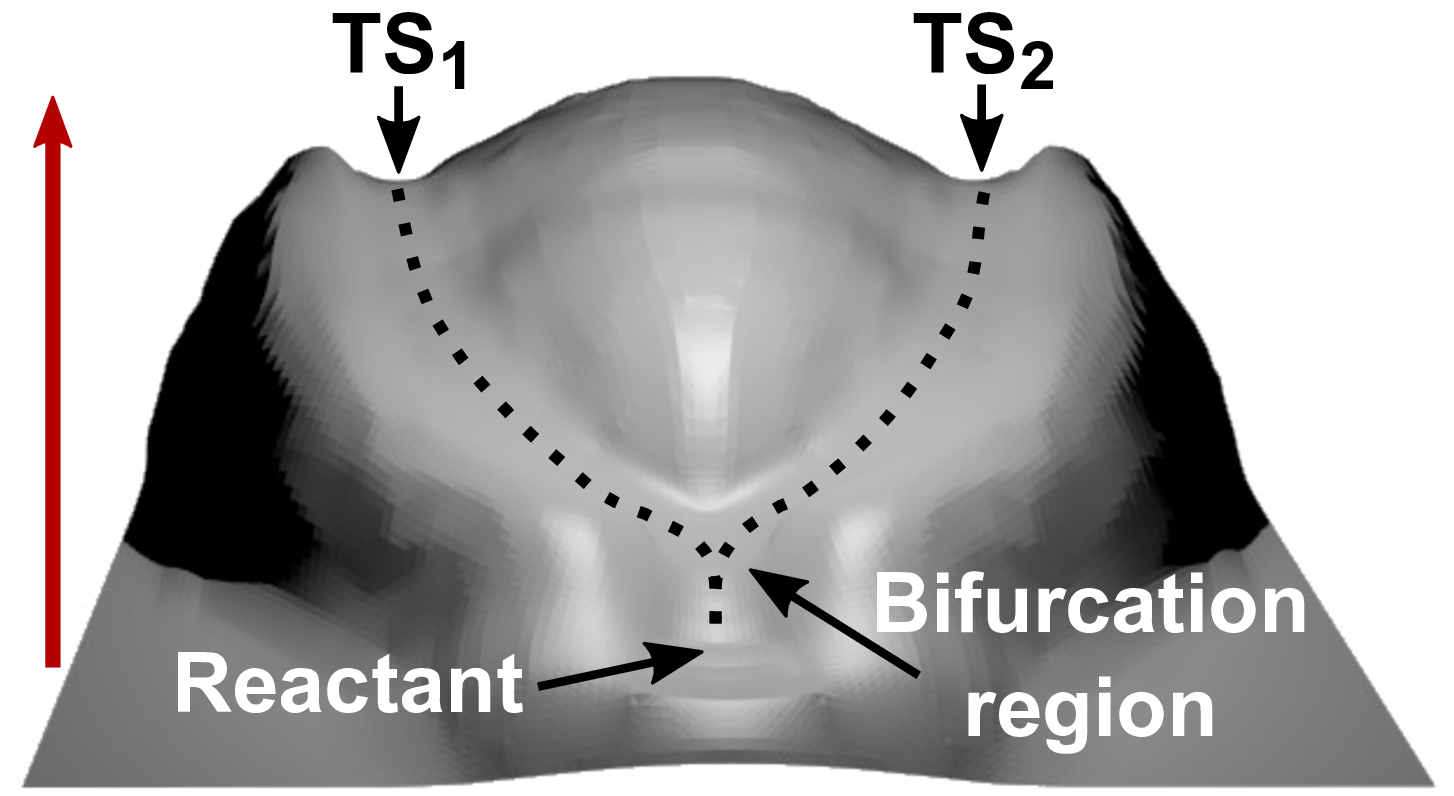
abstract = {The mechanochemistry of ring-opening reactions of cyclopropane derivatives turns out to be unexpectedly rich and puzzling. After showing that a rare so-called uphill bifurcation in the case of trans-gem-difluorocyclopropane turns into a downhill bifurcation upon substitution of fluorine by chlorine, bromine, and iodine in the thermal activation limit, the dichloro derivative is studied systematically in the realm of mechanochemical activation. Detailed exploration of the force-transformed potential energy surface of trans-gem-dichlorocyclopropane in terms of Dijkstra path analysis unveils a hitherto unknown topological catastrophe where the global shape of the energy landscape is fundamentally changed. From thermal activation up to moderately large forces, it is an uphill bifurcation that decides about dis- versus conrotatory ring-opening followed by separate transition states along both pathways. Above a critical force, the two distinct transition states merge to yield a single transition state such that the decision about the dis- versus conrotatory ring-opening process is taken at a newly established downhill bifurcation. The discovery of a force-induced qualitative change of the topology of a reaction network vastly transcends the previous understanding of the ring-opening reaction of this species. It would be astonishing to not discover a wealth of such catastrophes for mechanochemically activated reactions, which will greatly extend the known opportunities to manipulate chemical reaction networks.},
keywords = {Ab-Initio Molecular Dynamics (AIMD), Potential Energy Surface},
pubstate = {published},
tppubtype = {article}
}