2024
Pavan Ravindra, Xavier R. Advincula, Christoph Schran, Angelos Michaelides, Venkat Kapil
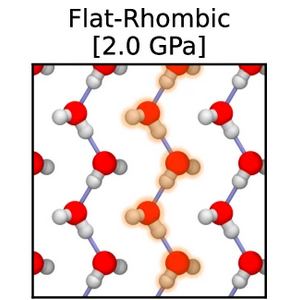
Quasi-one-dimensional hydrogen bonding in nanoconfined ice Journal Article
In: Nat. Commun., vol. 15, no. 1, pp. 1–9, 2024, ISSN: 20411723.
Abstract | Links | BibTeX | Tags: Confinement, Hydrogen bonding, Machine Learning Potentials, Water
@article{Ravindra2024/10.1038/s41467-024-51124-z,
title = {Quasi-one-dimensional hydrogen bonding in nanoconfined ice},
author = {Pavan Ravindra and Xavier R. Advincula and Christoph Schran and Angelos Michaelides and Venkat Kapil},
url = {https://www.nature.com/articles/s41467-024-51124-z},
doi = {10.1038/s41467-024-51124-z},
issn = {20411723},
year = {2024},
date = {2024-08-24},
urldate = {2024-08-24},
journal = {Nat. Commun.},
volume = {15},
number = {1},
pages = {1–9},
publisher = {Nature Publishing Group},
abstract = {The Bernal-Fowler ice rules stipulate that each water molecule in an ice crystal should form four hydrogen bonds. However, in extreme or constrained conditions, the arrangement of water molecules deviates from conventional ice rules, resulting in properties significantly different from bulk water. In this study, we employ machine learning-driven first-principles simulations to identify a new stabilization mechanism in nanoconfined ice phases. Instead of forming four hydrogen bonds, nanoconfined crystalline ice can form a quasi-one-dimensional hydrogen-bonded structure that exhibits only two hydrogen bonds per water molecule. These structures consist of strongly hydrogen-bonded linear chains of water molecules that zig-zag along one dimension, stabilized by van der Waals interactions that stack these chains along the other dimension. The unusual interplay of hydrogen bonding and van der Waals interactions in nanoconfined ice results in atypical proton behavior such as potential ferroelectric behavior, low dielectric response, and long-range proton dynamics.},
keywords = {Confinement, Hydrogen bonding, Machine Learning Potentials, Water},
pubstate = {published},
tppubtype = {article}
}
Kara D. Fong, Barbara Sumić, Niamh O’Neill, Christoph Schran, Clare P. Grey, Angelos Michaelides
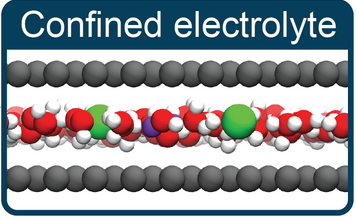
The Interplay of Solvation and Polarization Effects on Ion Pairing in Nanoconfined Electrolytes Journal Article
In: Nano Letters, vol. 24, no. 16, pp. 5024–5030, 2024, (PMID: 38592099).
Abstract | Links | BibTeX | Tags: Confinement, Hydrogen bonding, Ions in Water, Machine Learning Potentials
@article{Fong/10.1021/acs.nanolett.4c00890,
title = {The Interplay of Solvation and Polarization Effects on Ion Pairing in Nanoconfined Electrolytes},
author = {Kara D. Fong and Barbara Sumić and Niamh O’Neill and Christoph Schran and Clare P. Grey and Angelos Michaelides},
url = {https://doi.org/10.1021/acs.nanolett.4c00890},
doi = {10.1021/acs.nanolett.4c00890},
year = {2024},
date = {2024-04-09},
urldate = {2024-04-09},
journal = {Nano Letters},
volume = {24},
number = {16},
pages = {5024–5030},
abstract = {The nature of ion–ion interactions in electrolytes confined to nanoscale pores has important implications for energy storage and separation technologies. However, the physical effects dictating the structure of nanoconfined electrolytes remain debated. Here we employ machine-learning-based molecular dynamics simulations to investigate ion–ion interactions with density functional theory level accuracy in a prototypical confined electrolyte, aqueous NaCl within graphene slit pores. We find that the free energy of ion pairing in highly confined electrolytes deviates substantially from that in bulk solutions, observing a decrease in contact ion pairing but an increase in solvent-separated ion pairing. These changes arise from an interplay of ion solvation effects and graphene’s electronic structure. Notably, the behavior observed from our first-principles-level simulations is not reproduced even qualitatively with the classical force fields conventionally used to model these systems. The insight provided in this work opens new avenues for predicting and controlling the structure of nanoconfined electrolytes.},
note = {PMID: 38592099},
keywords = {Confinement, Hydrogen bonding, Ions in Water, Machine Learning Potentials},
pubstate = {published},
tppubtype = {article}
}
Thomas Dufils, Christoph Schran, Ji Chen, Andre K. Geim, Laura Fumagalli, Angelos Michaelides
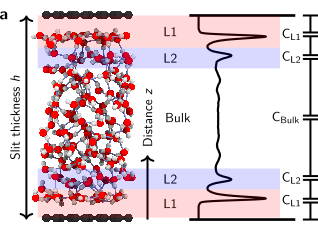
Origin of dielectric polarization suppression in confined water from first principles Journal Article
In: Chem. Sci., vol. 15, iss. 2, pp. 516–527, 2024.
Abstract | Links | BibTeX | Tags: AIMD, Confinement, Hydrogen bonding, Water
@article{Dufils2024/10.1039/D3SC04740G,
title = {Origin of dielectric polarization suppression in confined water from first principles},
author = {Thomas Dufils and Christoph Schran and Ji Chen and Andre K. Geim and Laura Fumagalli and Angelos Michaelides},
url = {http://dx.doi.org/10.1039/D3SC04740G},
doi = {10.1039/D3SC04740G},
year = {2024},
date = {2024-01-01},
urldate = {2024-01-01},
journal = {Chem. Sci.},
volume = {15},
issue = {2},
pages = {516–527},
publisher = {The Royal Society of Chemistry},
abstract = {It has long been known that the dielectric constant of confined water should be different from that in bulk. Recent experiments have shown that it is vanishingly small, however the origin of the phenomenon remains unclear. Here we used ab initio molecular dynamics simulations (AIMD) and AIMD-trained machine-learning potentials to understand water's structure and electronic properties underpinning this effect. For the graphene and hexagonal boron-nitride substrates considered, we find that it originates in the spontaneous anti-parallel alignment of the water dipoles in the first two water layers near the solid interface. The interfacial layers exhibit net ferroelectric ordering, resulting in an overall anti-ferroelectric arrangement of confined water. Together with constrained hydrogen-bonding orientations, this leads to much reduced out-of-plane polarization. Furthermore, we directly contrast AIMD and simple classical force-field simulations, revealing important differences. This work offers insight into a property of water that is critical in modulating surface forces, the electric-double-layer formation and molecular solvation, and shows a way to compute it.},
keywords = {AIMD, Confinement, Hydrogen bonding, Water},
pubstate = {published},
tppubtype = {article}
}
2022

Fabian L. Thiemann, Christoph Schran, Patrick Rowe, Erich A. Müller, Angelos Michaelides
Water flow in single-wall nanotubes: Oxygen makes it slip, hydrogen makes it stick Journal Article
In: ACS Nano, vol. 16, no. 7, pp. 10775–10782, 2022.
Abstract | Links | BibTeX | Tags: Confinement, Hydrogen bonding, Machine Learning Potentials, Water at Interfaces, Water flow
@article{Thiemann2022/10.1021/acsnano.2c02784,
title = {Water flow in single-wall nanotubes: Oxygen makes it slip, hydrogen makes it stick},
author = {Fabian L. Thiemann and Christoph Schran and Patrick Rowe and Erich A. Müller and Angelos Michaelides},
doi = {10.1021/acsnano.2c02784},
year = {2022},
date = {2022-06-01},
urldate = {2022-06-01},
journal = {ACS Nano},
volume = {16},
number = {7},
pages = {10775–10782},
abstract = {Experimental measurements have reported ultra-fast and radius-dependent water transport in carbon nanotubes which are absent in boron nitride nanotubes. Despite considerable effort, the origin of this contrasting (and fascinating) behaviour is not understood. Here, with the aid of machine learning-based molecular dynamics simulations that deliver first-principles accuracy, we investigate water transport in single-wall carbon and boron nitride nanotubes. Our simulations reveal a large, radius-dependent hydrodynamic slippage on both materials with water experiencing indeed a $backslashapprox 5$ times lower friction on carbon surfaces compared to boron nitride. Analysis of the diffusion mechanisms across the two materials reveals that the fast water transport on carbon is governed by facile oxygen motion, whereas the higher friction on boron nitride arises from specific hydrogen-nitrogen interactions. This work not only delivers a clear reference of unprecedented accuracy for water flow in single-wall nanotubes, but also provides detailed mechanistic insight into its radius and material dependence for future technological application.},
keywords = {Confinement, Hydrogen bonding, Machine Learning Potentials, Water at Interfaces, Water flow},
pubstate = {published},
tppubtype = {article}
}
2021
Christoph Schran, Fabien Brieuc, Dominik Marx
Transferability of machine learning potentials: Protonated water neural network potential applied to the protonated water hexamer Journal Article
In: J. Chem. Phys., vol. 154, no. 5, pp. 051101, 2021, ISSN: 10897690.
Abstract | Links | BibTeX | Tags: Coupled Cluster, Hydrogen bonding, Machine Learning Potentials, Water
@article{Schran2021/10.1063/5.0035438,
title = {Transferability of machine learning potentials: Protonated water neural network potential applied to the protonated water hexamer},
author = {Christoph Schran and Fabien Brieuc and Dominik Marx},
doi = {10.1063/5.0035438},
issn = {10897690},
year = {2021},
date = {2021-02-01},
urldate = {2021-02-01},
journal = {J. Chem. Phys.},
volume = {154},
number = {5},
pages = {051101},
abstract = {A previously published neural network potential for the description of protonated water clusters up to the protonated water tetramer, H+(H2O)4, at an essentially converged coupled cluster accuracy [C. Schran, J. Behler, and D. Marx, J. Chem. Theory Comput. 16, 88 (2020)] is applied to the protonated water hexamer, H+(H2O)6 - a system that the neural network has never seen before. Although being in the extrapolation regime, it is shown that the potential not only allows for quantum simulations from ultra-low temperatures ∼1 K up to 300 K but is also able to describe the new system very accurately compared to explicit coupled cluster calculations. This transferability of the model is rationalized by the similarity of the atomic environments encountered for the larger cluster compared to the environments in the training set of the model. Compared to the interpolation regime, the quality of the model is reduced by roughly one order of magnitude, but most of the difference to the coupled cluster reference comes from global shifts of the potential energy surface, while local energy fluctuations are well recovered. These results suggest that the application of neural network potentials in extrapolation regimes can provide useful results and might be more general than usually thought.},
keywords = {Coupled Cluster, Hydrogen bonding, Machine Learning Potentials, Water},
pubstate = {published},
tppubtype = {article}
}
2020
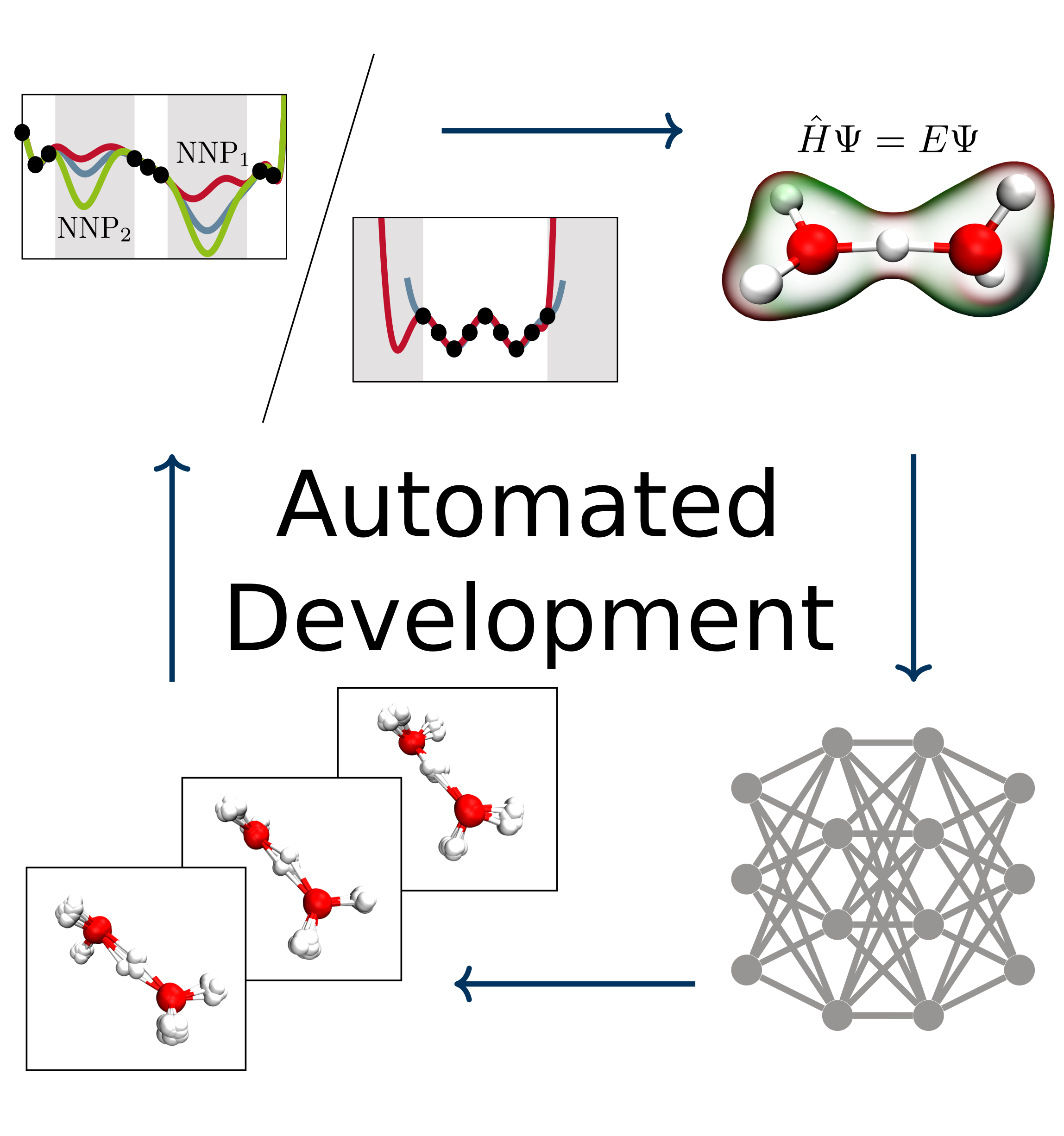
Christoph Schran, Jörg Behler, Dominik Marx
Automated Fitting of Neural Network Potentials at Coupled Cluster Accuracy: Protonated Water Clusters as Testing Ground Journal Article
In: J. Chem. Theory Comput., vol. 16, no. 1, pp. 88–99, 2020, ISSN: 15499626.
Abstract | Links | BibTeX | Tags: Coupled Cluster, Hydrogen bonding, Machine Learning Potentials, Water
@article{Schran2020/10.1021/acs.jctc.9b00805,
title = {Automated Fitting of Neural Network Potentials at Coupled Cluster Accuracy: Protonated Water Clusters as Testing Ground},
author = {Christoph Schran and Jörg Behler and Dominik Marx},
doi = {10.1021/acs.jctc.9b00805},
issn = {15499626},
year = {2020},
date = {2020-01-01},
urldate = {2020-01-01},
journal = {J. Chem. Theory Comput.},
volume = {16},
number = {1},
pages = {88–99},
abstract = {Highly accurate potential energy surfaces are of key interest for the detailed understanding and predictive modeling of chemical systems. In recent years, several new types of force fields, which are based on machine learning algorithms and fitted to ab initio reference calculations, have been introduced to meet this requirement. Here, we show how high-dimensional neural network potentials can be employed to automatically generate the potential energy surface of finite sized clusters at coupled cluster accuracy, namely CCSD(T*)-F12a/aug-cc-pVTZ. The developed automated procedure utilizes the established intrinsic properties of the model such that the configurations for the training set are selected in an unbiased and efficient way to minimize the computational effort of expensive reference calculations. These ideas are applied to protonated water clusters from the hydronium cation, H3O+, up to the tetramer, H9O4+, and lead to a single potential energy surface that describes all these systems at essentially converged coupled cluster accuracy with a fitting error of 0.06 kJ/mol per atom. The fit is validated in detail for all clusters up to the tetramer and yields reliable results not only for stationary points but also for reaction pathways and intermediate configurations as well as different sampling techniques. Per design, the neural network potentials (NNPs) constructed in this fashion can handle very different conditions including the quantum nature of the nuclei and enhanced sampling techniques covering very low as well as high temperatures. This enables fast and exhaustive exploration of the targeted protonated water clusters with essentially converged interactions. In addition, the automated process will allow one to tackle finite systems much beyond the present case.},
keywords = {Coupled Cluster, Hydrogen bonding, Machine Learning Potentials, Water},
pubstate = {published},
tppubtype = {article}
}
2019
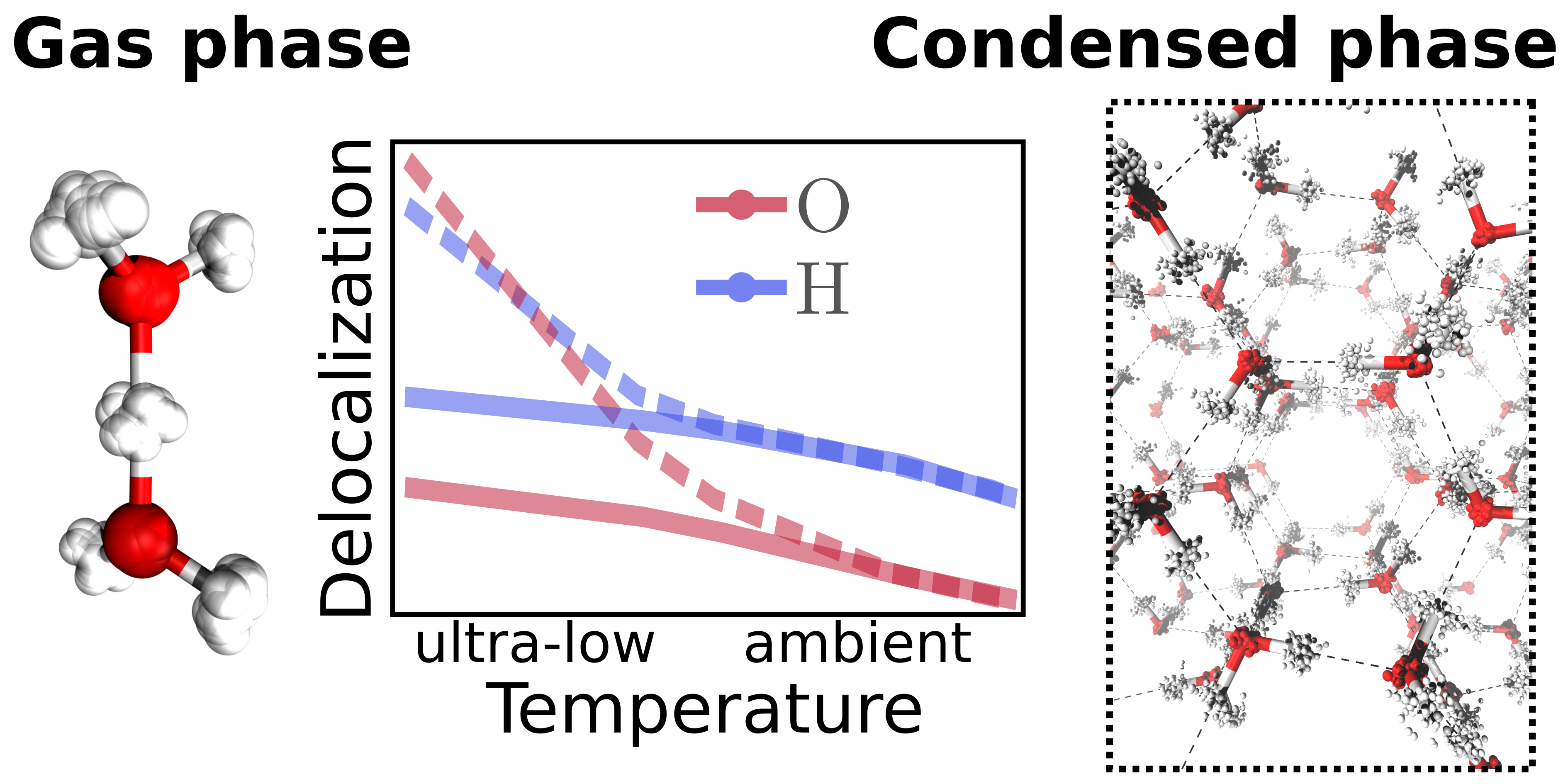
Christoph Schran, Dominik Marx
Quantum nature of the hydrogen bond from ambient conditions down to ultra-low temperatures Journal Article
In: Phys. Chem. Chem. Phys., vol. 21, no. 45, pp. 24967–24975, 2019, ISSN: 14639076.
Abstract | Links | BibTeX | Tags: Hydrogen bonding, Nuclear quantum effects, path integral molecular dynamics (PIMD), Water
@article{Schran2019/10.1039/C9CP04795F,
title = {Quantum nature of the hydrogen bond from ambient conditions down to ultra-low temperatures},
author = {Christoph Schran and Dominik Marx},
doi = {10.1039/c9cp04795f},
issn = {14639076},
year = {2019},
date = {2019-10-01},
urldate = {2019-10-01},
journal = {Phys. Chem. Chem. Phys.},
volume = {21},
number = {45},
pages = {24967–24975},
abstract = {Many experimental techniques such as tagging photodissociation and helium nanodroplet isolation spectroscopy operate at very low temperatures in order to investigate hydrogen bonding. To elucidate the differences between such ultra-cold and usual ambient conditions, different hydrogen bonded systems are studied systematically from 300 K down to about 1 K using path integral simulations that explicitly consider both the quantum nature of the nuclei and thermal fluctuations. For this purpose, finite sized water clusters, specifically the water dimer and hexamer, protonated water clusters including the Zundel and Eigen complexes, as well as hexagonal ice as a condensed phase representative are compared directly as a function of temperature. While weaker hydrogen bonds, as present in the neutral systems, show distinct structural differences between ambient conditions and the ultra-cold regime, the stronger hydrogen bonds of the protonated water clusters are less perturbed by temperature compared to their quantum ground state. In all the studied systems, the quantum delocalization of the nuclei is found to vary drastically with temperature. Interestingly, upon reaching temperatures of about 1 K, the spatial quantum delocalization of the heavy oxygens approaches that of the protons for relatively weak spatial constraints, and even significantly exceeds the latter in the case of the centered hydrogen bond in the Zundel complex. These findings are relevant for comparisons between experiments on hydrogen bonding carried out under ultra-cold versus ambient conditions as well as to understand quantum delocalization phenomena of nuclei by seamlessly extending our insights into noncovalent interactions down to ultra-low temperatures.},
keywords = {Hydrogen bonding, Nuclear quantum effects, path integral molecular dynamics (PIMD), Water},
pubstate = {published},
tppubtype = {article}
}
2017
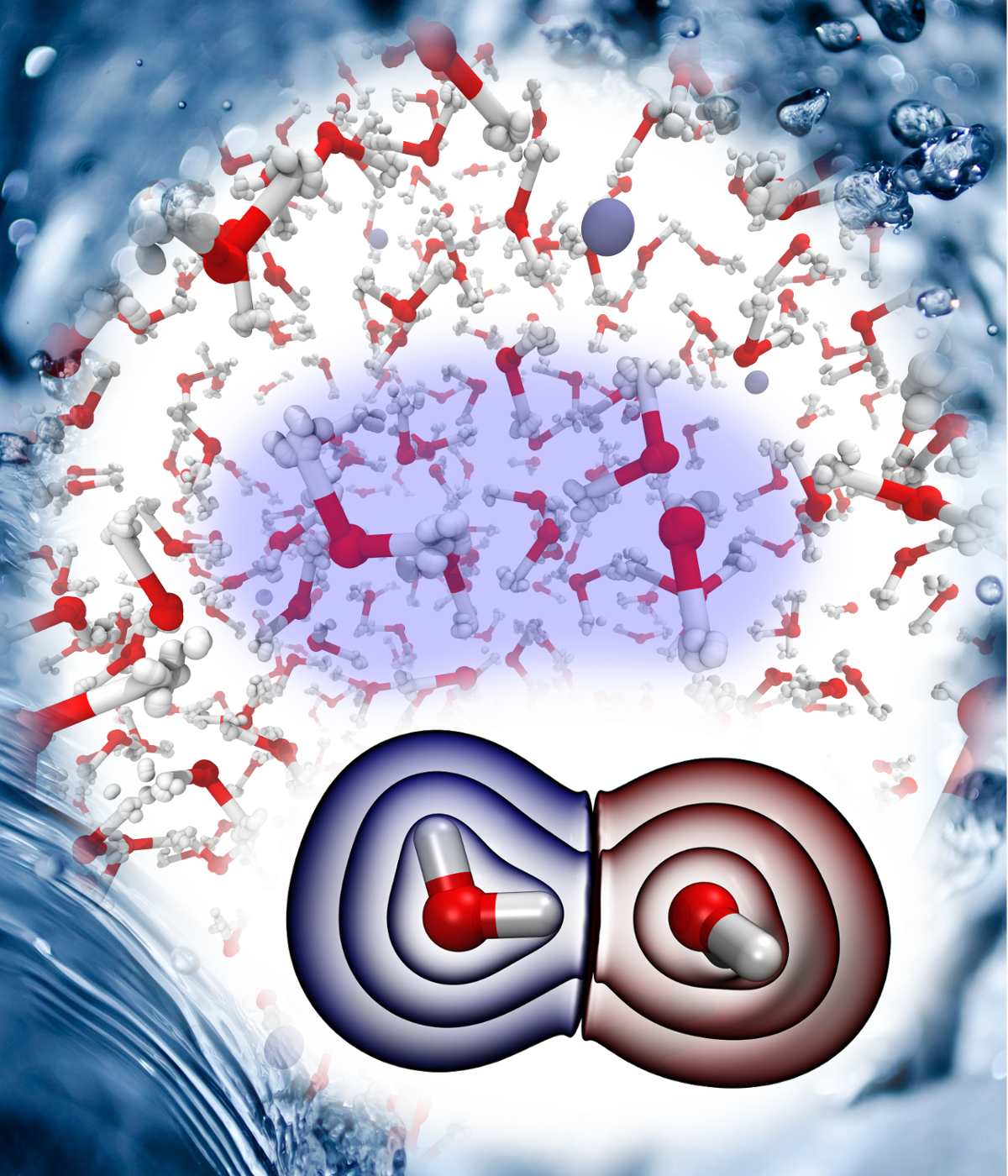
Christoph Schran, Ondrej Marsalek, Thomas E. Markland
Unravelling the influence of quantum proton delocalization on electronic charge transfer through the hydrogen bond Journal Article
In: Chem. Phys. Lett., vol. 678, pp. 289–295, 2017, ISSN: 00092614.
Abstract | Links | BibTeX | Tags: Charge transfer, Hydrogen bonding, Ions in Water, Nuclear quantum effects, Water
@article{Schran2017/10.1016/j.cplett.2017.04.034,
title = {Unravelling the influence of quantum proton delocalization on electronic charge transfer through the hydrogen bond},
author = {Christoph Schran and Ondrej Marsalek and Thomas E. Markland},
doi = {10.1016/j.cplett.2017.04.034},
issn = {00092614},
year = {2017},
date = {2017-06-01},
urldate = {2017-06-01},
journal = {Chem. Phys. Lett.},
volume = {678},
pages = {289–295},
abstract = {Upon hydrogen bond formation, electronic charge density is transferred between the donor and acceptor, impacting processes ranging from hydration to spectroscopy. Here we use ab initio path integral simulations to elucidate the role of nuclear quantum effects in determining the charge transfer in a range of hydrogen bonded species in the gas and liquid phase. We show that the quantization of the nuclei gives rise to large changes in the magnitude of the charge transfer as well as its temperature dependence. We then explain how a single geometric parameter determines the charge transfer through the hydrogen bond. These results thus demonstrate that nuclear quantum effects are vital for the accurate description of charge transfer and offer a physically transparent way to understand how hydrogen bonding gives rise to it.},
keywords = {Charge transfer, Hydrogen bonding, Ions in Water, Nuclear quantum effects, Water},
pubstate = {published},
tppubtype = {article}
}
2016
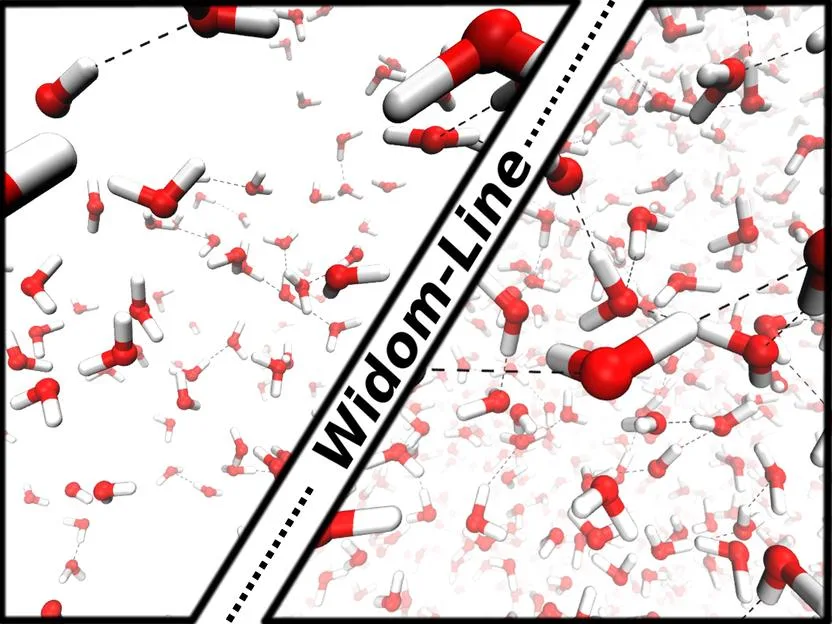
Maciej Śmiechowski, Christoph Schran, Harald Forbert, Dominik Marx
Correlated Particle Motion and THz Spectral Response of Supercritical Water Journal Article
In: Phys. Rev. Lett., vol. 116, no. 2, 2016, ISSN: 10797114.
Abstract | Links | BibTeX | Tags: Hydrogen bonding, Spectra, Water
@article{Smiechowski2016/10.1103/PhysRevLett.116.027801,
title = {Correlated Particle Motion and THz Spectral Response of Supercritical Water},
author = {Maciej Śmiechowski and Christoph Schran and Harald Forbert and Dominik Marx},
doi = {10.1103/PhysRevLett.116.027801},
issn = {10797114},
year = {2016},
date = {2016-01-01},
urldate = {2016-01-01},
journal = {Phys. Rev. Lett.},
volume = {116},
number = {2},
abstract = {Molecular dynamics simulations of supercritical water reveal distinctly different distance-dependent modulations of dipolar response and correlations in particle motion compared to ambient conditions. The strongly perturbed H-bond network of water at supercritical conditions allows for considerable translational and rotational freedom of individual molecules. These changes give rise to substantially different infrared spectra and vibrational density of states at THz frequencies for densities above and below the Widom line that separates percolating liquidlike and clustered gaslike supercritical water.},
keywords = {Hydrogen bonding, Spectra, Water},
pubstate = {published},
tppubtype = {article}
}