2023
Fabien Brieuc, Christoph Schran, Dominik Marx
Manifestations of local supersolidity of $^4$He around a charged molecular impurity Journal Article
In: Phys. Rev. Res., vol. 5, iss. 4, pp. 043083, 2023.
Links | BibTeX | Tags: Nuclear quantum effects, path integral molecular dynamics (PIMD), Superfluidity
@article{PhysRevResearch.5.043083,
title = {Manifestations of local supersolidity of $^4$He around a charged molecular impurity},
author = {Fabien Brieuc and Christoph Schran and Dominik Marx},
url = {https://link.aps.org/doi/10.1103/PhysRevResearch.5.043083},
doi = {10.1103/PhysRevResearch.5.043083},
year = {2023},
date = {2023-10-01},
urldate = {2023-10-01},
journal = {Phys. Rev. Res.},
volume = {5},
issue = {4},
pages = {043083},
publisher = {American Physical Society},
keywords = {Nuclear quantum effects, path integral molecular dynamics (PIMD), Superfluidity},
pubstate = {published},
tppubtype = {article}
}
Julia A. Davies, Christoph Schran, Fabien Brieuc, Dominik Marx, Andrew M. Ellis
Onset of Rotational Decoupling for a Molecular Ion Solvated in Helium: From Tags to Rings and Shells Journal Article
In: Phys. Rev. Lett., vol. 130, iss. 8, pp. 083001, 2023.
Links | BibTeX | Tags: Nuclear quantum effects, path integral molecular dynamics (PIMD), Superfluidity, Water
@article{Schran2023/10.1103/PhysRevLett.130.083001,
title = {Onset of Rotational Decoupling for a Molecular Ion Solvated in Helium: From Tags to Rings and Shells},
author = {Julia A. Davies and Christoph Schran and Fabien Brieuc and Dominik Marx and Andrew M. Ellis},
doi = {10.1103/PhysRevLett.130.083001},
year = {2023},
date = {2023-02-01},
urldate = {2023-02-01},
journal = {Phys. Rev. Lett.},
volume = {130},
issue = {8},
pages = {083001},
publisher = {American Physical Society},
keywords = {Nuclear quantum effects, path integral molecular dynamics (PIMD), Superfluidity, Water},
pubstate = {published},
tppubtype = {article}
}
Irén Simkó, Christoph Schran, Fabien Brieuc, Csaba Fábri, Oskar Asvany, Stephan Schlemmer, Dominik Marx, Attila G. Császár
Quantum Nuclear Delocalization and its Rovibrational Fingerprints Journal Article
In: Angewandte Chemie International Edition, vol. 62, no. 41, pp. e202306744, 2023.
Abstract | Links | BibTeX | Tags: Nuclear quantum effects, path integral molecular dynamics (PIMD), Quantum Dynamics
@article{Simko/10.1002/anie.202306744,
title = {Quantum Nuclear Delocalization and its Rovibrational Fingerprints},
author = {Irén Simkó and Christoph Schran and Fabien Brieuc and Csaba Fábri and Oskar Asvany and Stephan Schlemmer and Dominik Marx and Attila G. Császár},
url = {https://onlinelibrary.wiley.com/doi/abs/10.1002/anie.202306744},
doi = {10.1002/anie.202306744},
year = {2023},
date = {2023-01-01},
urldate = {2023-01-01},
journal = {Angewandte Chemie International Edition},
volume = {62},
number = {41},
pages = {e202306744},
abstract = {Abstract Quantum mechanics dictates that nuclei must undergo some delocalization. In this work, emergence of quantum nuclear delocalization and its rovibrational fingerprints are discussed for the case of the van der Waals complex . The equilibrium structure of is planar and T-shaped, one He atom solvating the quasi-linear He−H+−He core. The dynamical structure of , in all of its bound states, is fundamentally different. As revealed by spatial distribution functions and nuclear densities, during the vibrations of the molecule the solvating He is not restricted to be in the plane defined by the instantaneously bent chomophore, but freely orbits the central proton, forming a three-dimensional torus around the chromophore. This quantum delocalization is observed for all vibrational states, the type of vibrational excitation being reflected in the topology of the nodal surfaces in the nuclear densities, showing, for example, that intramolecular bending involves excitation along the circumference of the torus.},
keywords = {Nuclear quantum effects, path integral molecular dynamics (PIMD), Quantum Dynamics},
pubstate = {published},
tppubtype = {article}
}
2020
Christoph Schran, Krystof Brezina, Ondrej Marsalek
Committee neural network potentials control generalization errors and enable active learning Journal Article
In: J. Chem. Phys., vol. 153, no. 10, pp. 104105, 2020, ISSN: 10897690.
Abstract | Links | BibTeX | Tags: Machine Learning Potentials, path integral molecular dynamics (PIMD), Water
@article{Schran2020/10.1063/5.0016004,
title = {Committee neural network potentials control generalization errors and enable active learning},
author = {Christoph Schran and Krystof Brezina and Ondrej Marsalek},
doi = {10.1063/5.0016004},
issn = {10897690},
year = {2020},
date = {2020-09-01},
urldate = {2020-09-01},
journal = {J. Chem. Phys.},
volume = {153},
number = {10},
pages = {104105},
abstract = {It is well known in the field of machine learning that committee models improve accuracy, provide generalization error estimates, and enable active learning strategies. In this work, we adapt these concepts to interatomic potentials based on artificial neural networks. Instead of a single model, multiple models that share the same atomic environment descriptors yield an average that outperforms its individual members as well as a measure of the generalization error in the form of the committee disagreement. We not only use this disagreement to identify the most relevant configurations to build up the model's training set in an active learning procedure but also monitor and bias it during simulations to control the generalization error. This facilitates the adaptive development of committee neural network potentials and their training sets while keeping the number of ab initio calculations to a minimum. To illustrate the benefits of this methodology, we apply it to the development of a committee model for water in the condensed phase. Starting from a single reference ab initio simulation, we use active learning to expand into new state points and to describe the quantum nature of the nuclei. The final model, trained on 814 reference calculations, yields excellent results under a range of conditions, from liquid water at ambient and elevated temperatures and pressures to different phases of ice, and the air-water interface - all including nuclear quantum effects. This approach to committee models will enable the systematic development of robust machine learning models for a broad range of systems.},
keywords = {Machine Learning Potentials, path integral molecular dynamics (PIMD), Water},
pubstate = {published},
tppubtype = {article}
}
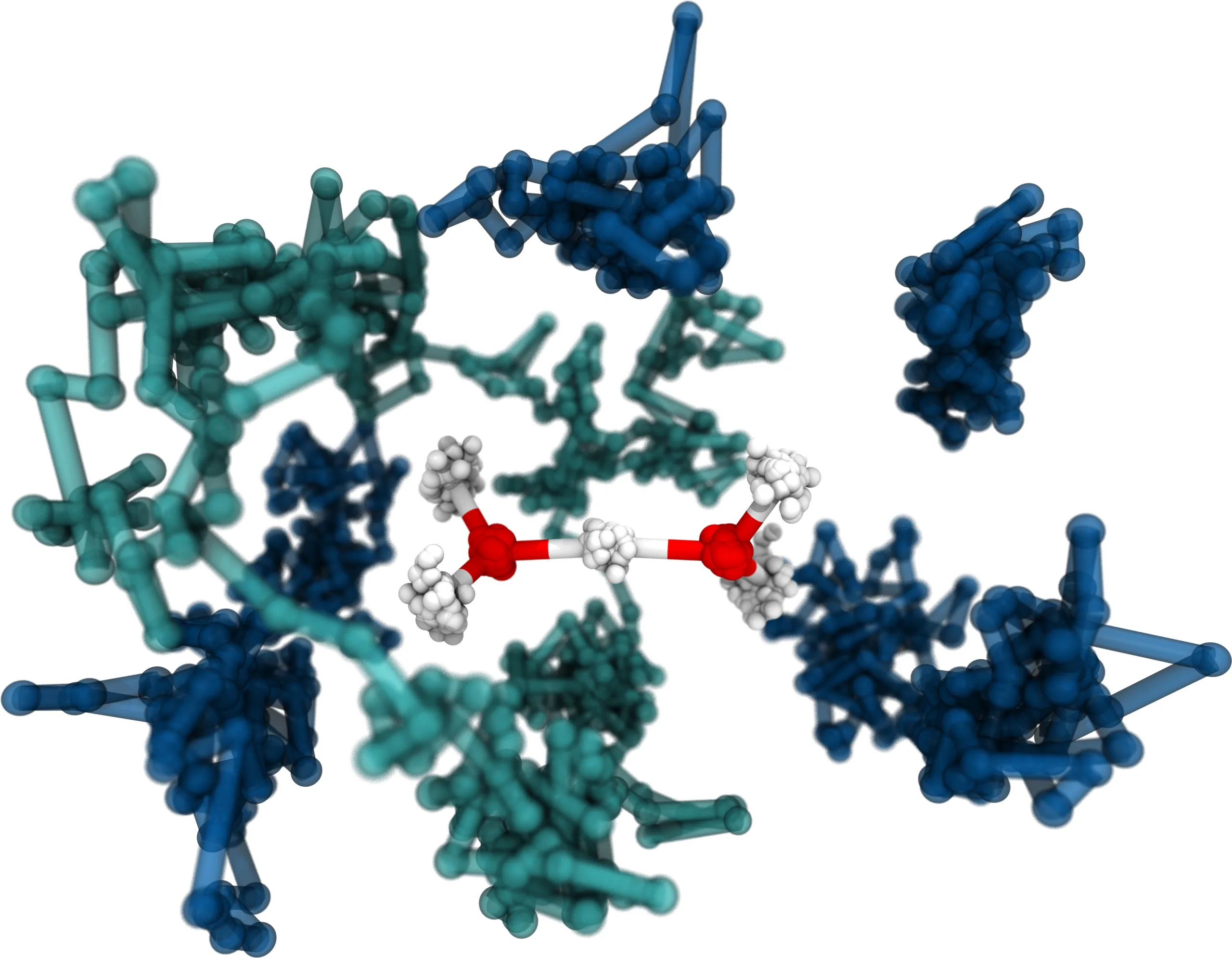
Fabien Brieuc, Christoph Schran, Felix Uhl, Harald Forbert, Dominik Marx
Converged quantum simulations of reactive solutes in superfluid helium: The Bochum perspective Journal Article
In: J. Chem. Phys., vol. 152, no. 21, pp. 210901, 2020, ISSN: 10897690.
Abstract | Links | BibTeX | Tags: Nuclear quantum effects, path integral molecular dynamics (PIMD), Superfluidity
@article{Brieuc2020/10.1063/5.0008309,
title = {Converged quantum simulations of reactive solutes in superfluid helium: The Bochum perspective},
author = {Fabien Brieuc and Christoph Schran and Felix Uhl and Harald Forbert and Dominik Marx},
doi = {10.1063/5.0008309},
issn = {10897690},
year = {2020},
date = {2020-06-01},
urldate = {2020-06-01},
journal = {J. Chem. Phys.},
volume = {152},
number = {21},
pages = {210901},
abstract = {Superfluid helium has not only fascinated scientists for centuries but is also the ideal matrix for the investigation of chemical systems under ultra-cold conditions in helium nanodroplet isolation experiments. Together with related experimental techniques such as helium tagging photodissociation spectroscopy, these methods have provided unique insights into many interesting systems. Complemented by theoretical work, they were additionally able to greatly expand our general understanding of manifestations of superfluid behavior in finite sized clusters and their response to molecular impurities. However, most theoretical studies up to now have not included the reactivity and flexibility of molecular systems embedded in helium. In this perspective, the theoretical foundation of simulating fluxional molecules and reactive complexes in superfluid helium is presented in detail. Special emphasis is put on recent developments for the converged description of both the molecular interactions and the quantum nature of the nuclei at ultra-low temperatures. As a first step, our hybrid path integral molecular dynamics/bosonic path integral Monte Carlo method is reviewed. Subsequently, methods for efficient path integral sampling tailored for this hybrid coupling scheme are discussed while also introducing new developments to enhance the accurate incorporation of the solute⋯solvent coupling. Finally, highly accurate descriptions of the interactions in solute⋯helium systems using machine learning techniques are addressed. Our current automated and adaptive fitting procedures to parameterize high-dimensional neural network potentials for both the full-dimensional potential energy surface of solutes and the solute⋯solvent interaction potentials are concisely presented. They are demonstrated to faithfully represent many-body potential functions able to describe chemically complex and reactive solutes in helium environments seamlessly from one He atom up to bulk helium at the accuracy level of coupled cluster electronic structure calculations. Together, these advances allow for converged quantum simulations of fluxional and reactive solutes in superfluid helium under cryogenic conditions.},
keywords = {Nuclear quantum effects, path integral molecular dynamics (PIMD), Superfluidity},
pubstate = {published},
tppubtype = {article}
}
2019
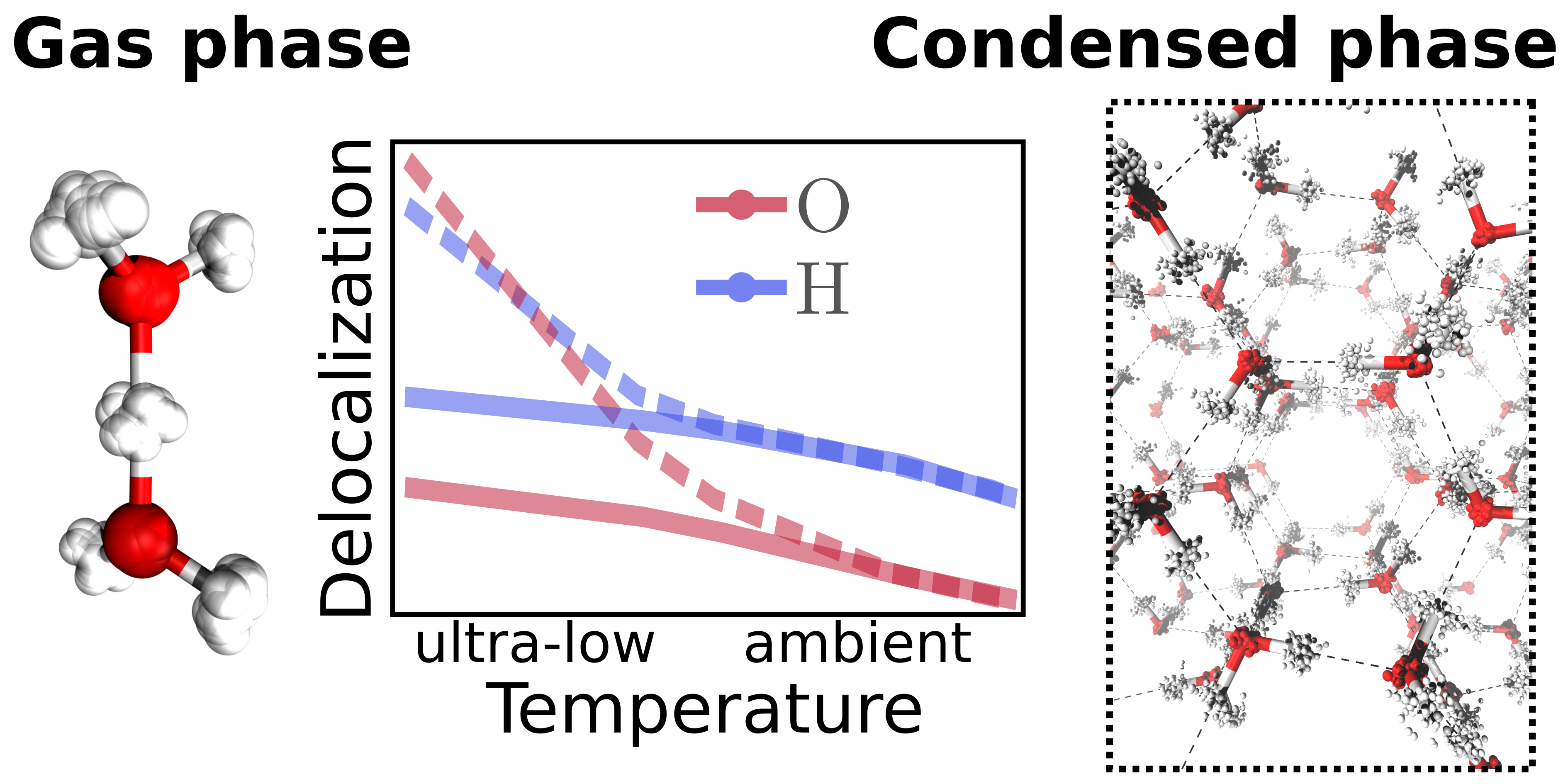
Christoph Schran, Dominik Marx
Quantum nature of the hydrogen bond from ambient conditions down to ultra-low temperatures Journal Article
In: Phys. Chem. Chem. Phys., vol. 21, no. 45, pp. 24967–24975, 2019, ISSN: 14639076.
Abstract | Links | BibTeX | Tags: Hydrogen bonding, Nuclear quantum effects, path integral molecular dynamics (PIMD), Water
@article{Schran2019/10.1039/C9CP04795F,
title = {Quantum nature of the hydrogen bond from ambient conditions down to ultra-low temperatures},
author = {Christoph Schran and Dominik Marx},
doi = {10.1039/c9cp04795f},
issn = {14639076},
year = {2019},
date = {2019-10-01},
urldate = {2019-10-01},
journal = {Phys. Chem. Chem. Phys.},
volume = {21},
number = {45},
pages = {24967–24975},
abstract = {Many experimental techniques such as tagging photodissociation and helium nanodroplet isolation spectroscopy operate at very low temperatures in order to investigate hydrogen bonding. To elucidate the differences between such ultra-cold and usual ambient conditions, different hydrogen bonded systems are studied systematically from 300 K down to about 1 K using path integral simulations that explicitly consider both the quantum nature of the nuclei and thermal fluctuations. For this purpose, finite sized water clusters, specifically the water dimer and hexamer, protonated water clusters including the Zundel and Eigen complexes, as well as hexagonal ice as a condensed phase representative are compared directly as a function of temperature. While weaker hydrogen bonds, as present in the neutral systems, show distinct structural differences between ambient conditions and the ultra-cold regime, the stronger hydrogen bonds of the protonated water clusters are less perturbed by temperature compared to their quantum ground state. In all the studied systems, the quantum delocalization of the nuclei is found to vary drastically with temperature. Interestingly, upon reaching temperatures of about 1 K, the spatial quantum delocalization of the heavy oxygens approaches that of the protons for relatively weak spatial constraints, and even significantly exceeds the latter in the case of the centered hydrogen bond in the Zundel complex. These findings are relevant for comparisons between experiments on hydrogen bonding carried out under ultra-cold versus ambient conditions as well as to understand quantum delocalization phenomena of nuclei by seamlessly extending our insights into noncovalent interactions down to ultra-low temperatures.},
keywords = {Hydrogen bonding, Nuclear quantum effects, path integral molecular dynamics (PIMD), Water},
pubstate = {published},
tppubtype = {article}
}
2018
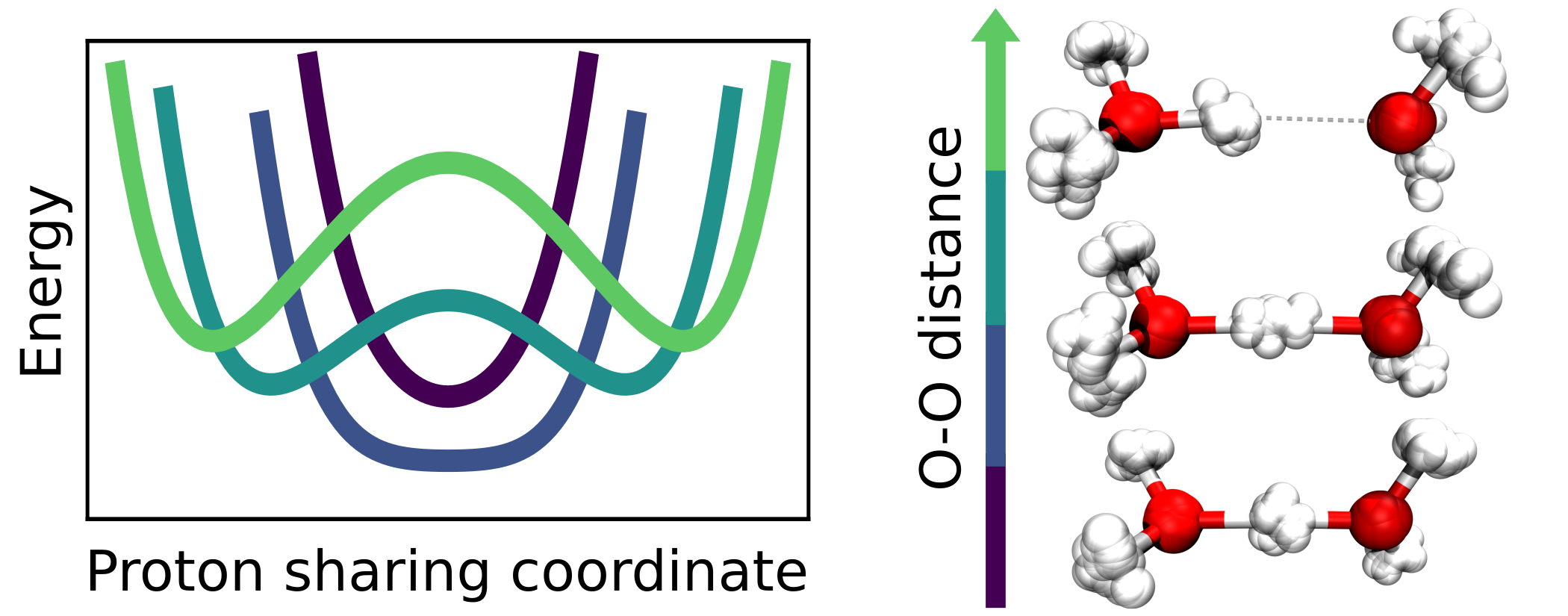
Christoph Schran, Fabien Brieuc, Dominik Marx
Converged Colored Noise Path Integral Molecular Dynamics Study of the Zundel Cation Down to Ultralow Temperatures at Coupled Cluster Accuracy Journal Article
In: J. Chem. Theory Comput., vol. 14, no. 10, pp. 5068–5078, 2018, ISSN: 15499626.
Abstract | Links | BibTeX | Tags: Nuclear quantum effects, path integral molecular dynamics (PIMD), Water
@article{Schran2018/10.1021/acs.jctc.8b00705,
title = {Converged Colored Noise Path Integral Molecular Dynamics Study of the Zundel Cation Down to Ultralow Temperatures at Coupled Cluster Accuracy},
author = {Christoph Schran and Fabien Brieuc and Dominik Marx},
doi = {10.1021/acs.jctc.8b00705},
issn = {15499626},
year = {2018},
date = {2018-09-01},
urldate = {2018-09-01},
journal = {J. Chem. Theory Comput.},
volume = {14},
number = {10},
pages = {5068–5078},
abstract = {For a long time, performing converged path integral simulations at ultra-low, but finite temperatures of a few Kelvin has been a nearly impossible task. However, recent developments in advanced colored noise thermostatting schemes for path integral simulations, namely the Path Integral Generalized Langevin Equation Thermostat (PIGLET) and the Path Integral Quantum Thermal Bath (PIQTB), have been able to greatly reduce the computational cost of these simulations, thus making the ultra-low temperature regime accessible in practice. In this work, we investigate the influence of these two thermostatting schemes on the description of hydrogen-bonded systems at temperatures down to a few Kelvin as encountered, for example, in helium nanodroplet isolation or tagging photodissociation spectroscopy experiments. For this purpose, we analyze the prototypical hydrogen bond in the Zundel cation (H5O2+) as a function of both, oxygen-oxygen distance and temperature in order to elucidate how the anisotropic quantum deloc...},
keywords = {Nuclear quantum effects, path integral molecular dynamics (PIMD), Water},
pubstate = {published},
tppubtype = {article}
}